

Ubiquitin ligase CHFR mediated degradation of VE-cadherin through ubiquitylation disrupts endothelial adherens junctions
- PMID: 37852964
- PMCID: PMC10584835
- DOI: 10.1038/s41467-023-42225-2
Ubiquitin ligase CHFR mediated degradation of VE-cadherin through ubiquitylation disrupts endothelial adherens junctions
Abstract
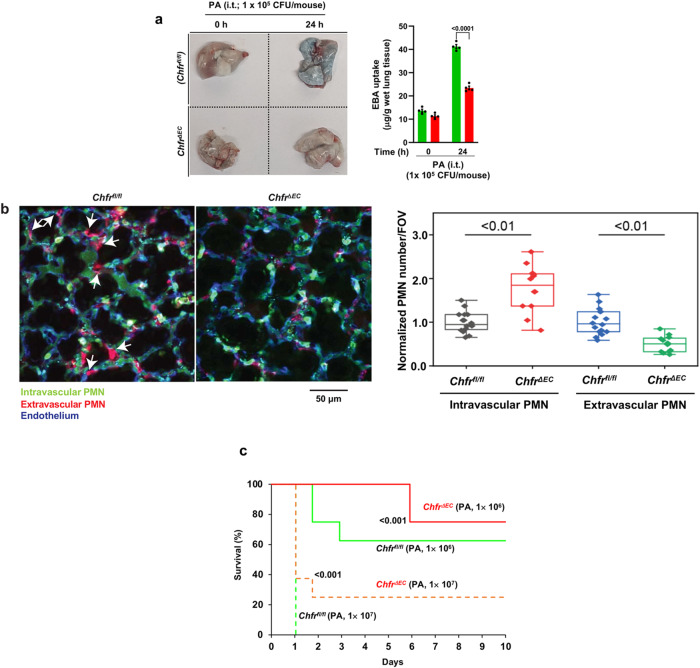
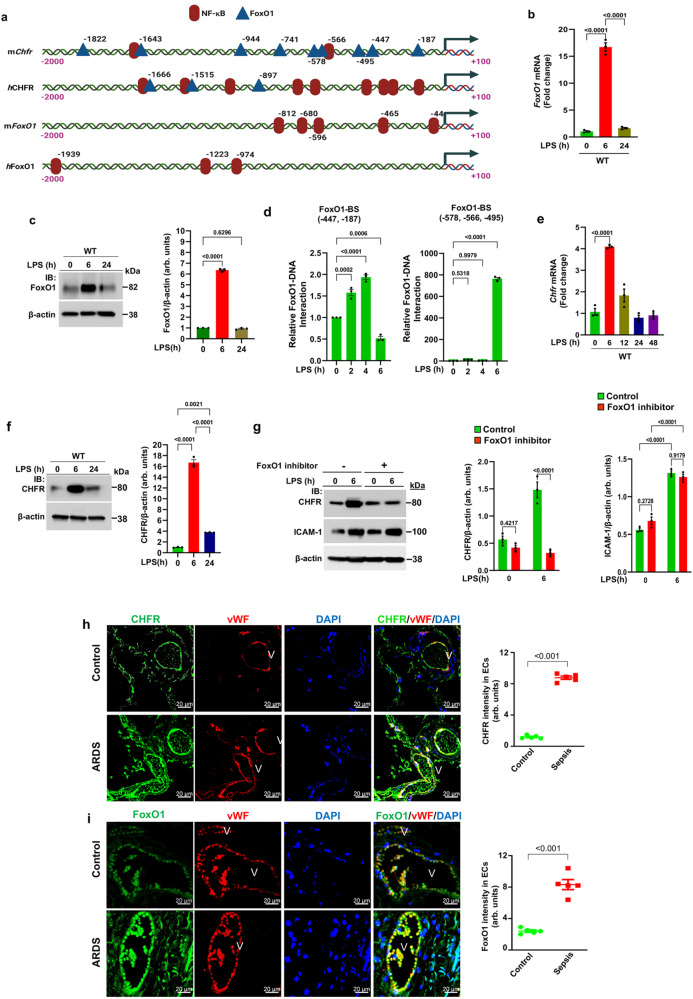
Vascular endothelial cadherin (VE-cadherin) expressed at endothelial adherens junctions (AJs) is vital for vascular integrity and endothelial homeostasis. Here we identify the requirement of the ubiquitin E3-ligase CHFR as a key mechanism of ubiquitylation-dependent degradation of VE-cadherin. CHFR was essential for disrupting the endothelium through control of the VE-cadherin protein expression at AJs. We observe augmented expression of VE-cadherin in endothelial cell (EC)-restricted Chfr knockout (ChfrΔEC) mice. We also observe abrogation of LPS-induced degradation of VE-cadherin in ChfrΔEC mice, suggesting the pathophysiological relevance of CHFR in regulating the endothelial junctional barrier in inflammation. Lung endothelial barrier breakdown, inflammatory neutrophil extravasation, and mortality induced by LPS were all suppressed in ChfrΔEC mice. We find that the transcription factor FoxO1 is a key upstream regulator of CHFR expression. These findings demonstrate the requisite role of the endothelial cell-expressed E3-ligase CHFR in regulating the expression of VE-cadherin, and thereby endothelial junctional barrier integrity.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
