

Chromatin compartmentalization regulates the response to DNA damage
- PMID: 37853125
- PMCID: PMC10620078
- DOI: 10.1038/s41586-023-06635-y
Chromatin compartmentalization regulates the response to DNA damage
Erratum in
-
Author Correction: Chromatin compartmentalization regulates the response to DNA damage.Nature. 2023 Dec;624(7990):E1. doi: 10.1038/s41586-023-06841-8. Nature. 2023. PMID: 37974007 Free PMC article. No abstract available.
Abstract
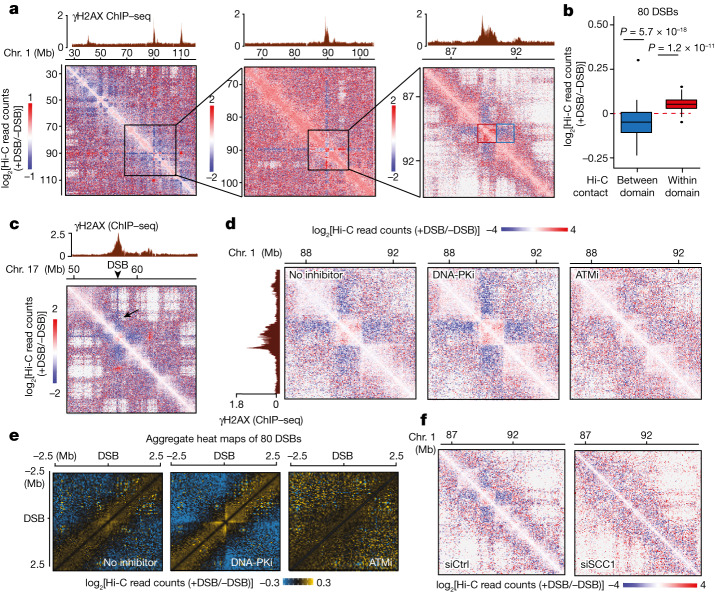
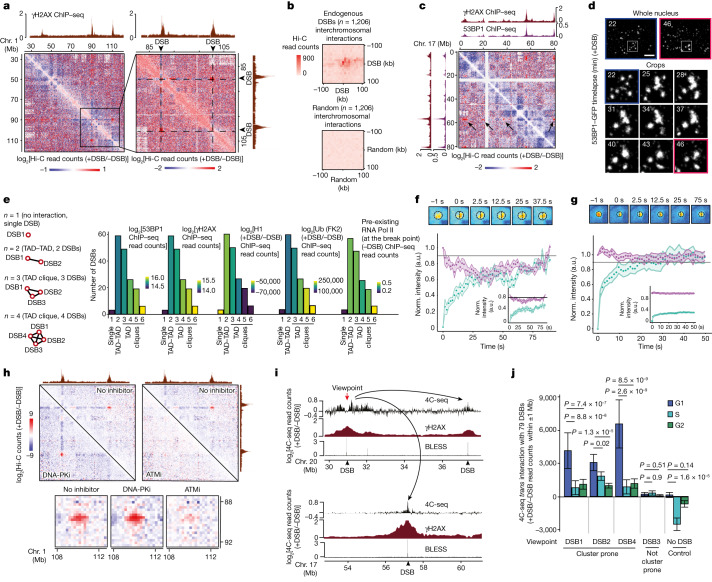
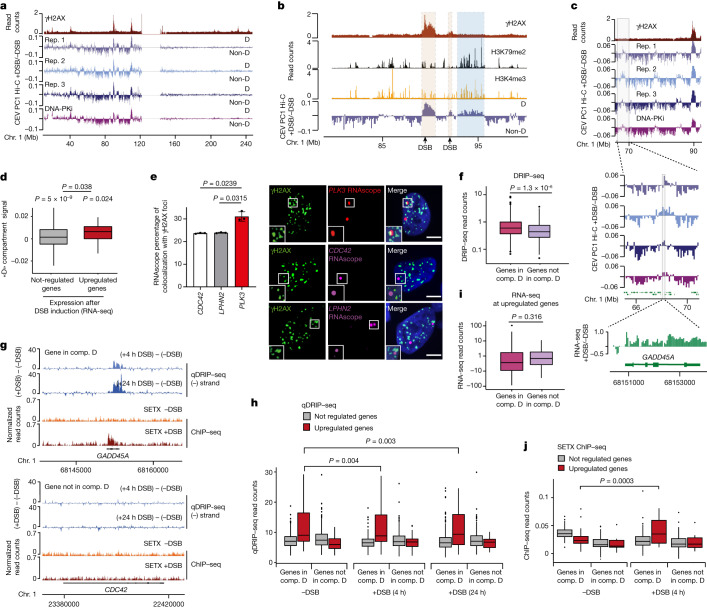
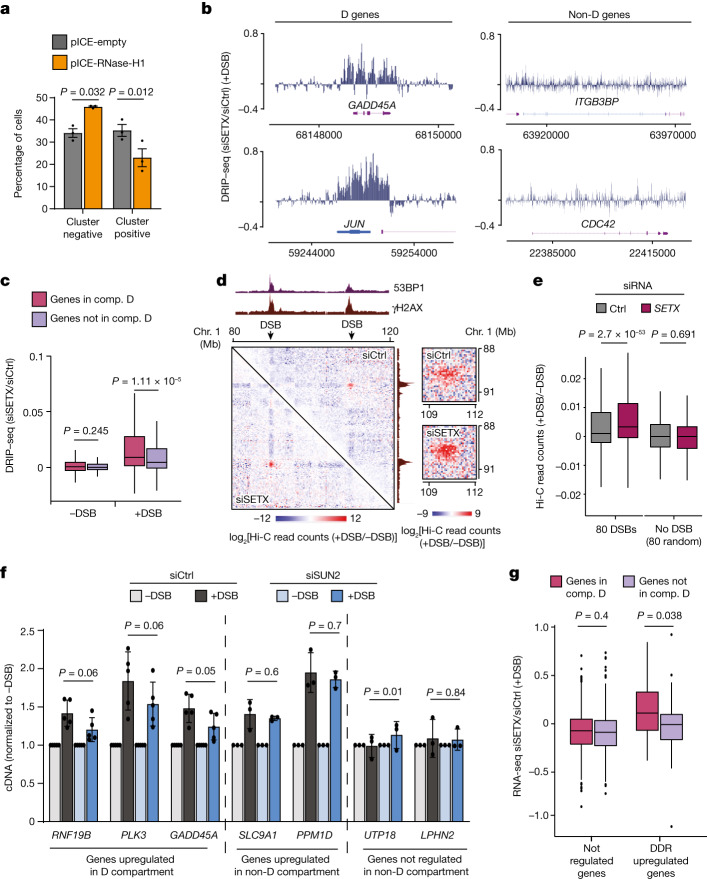
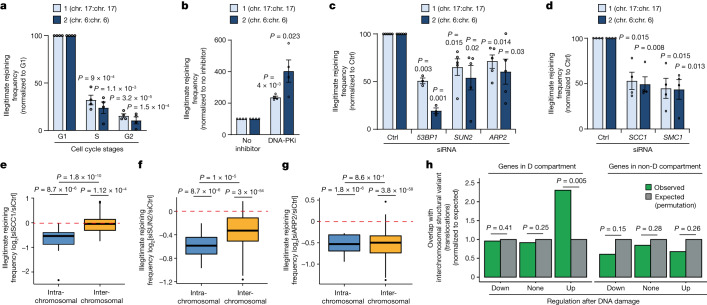
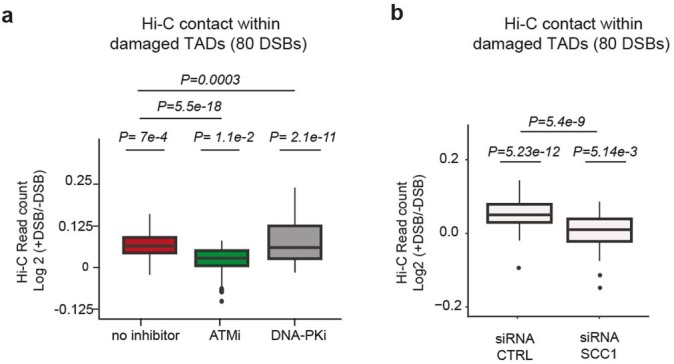
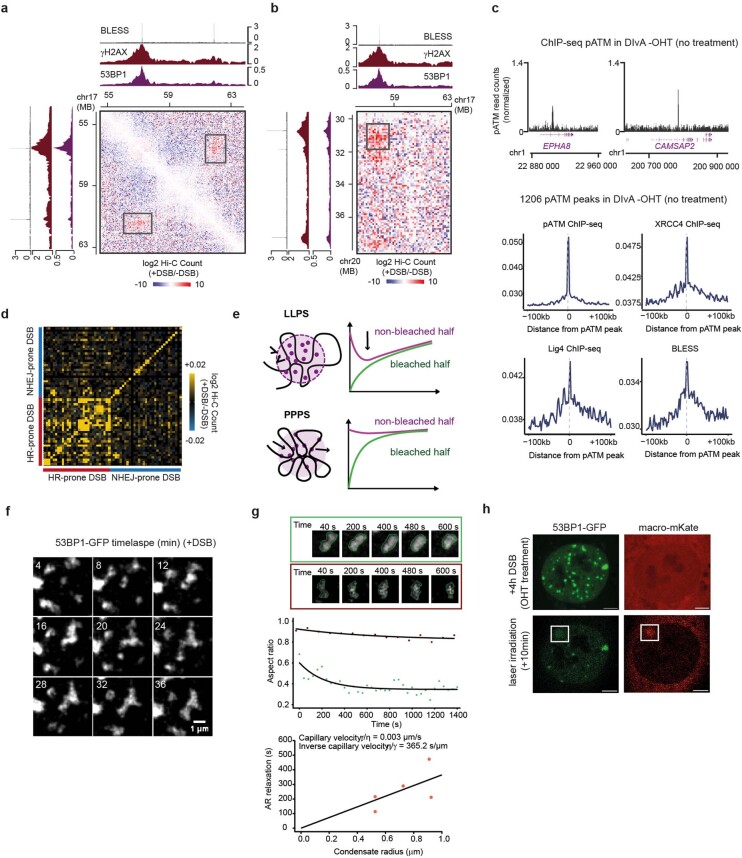
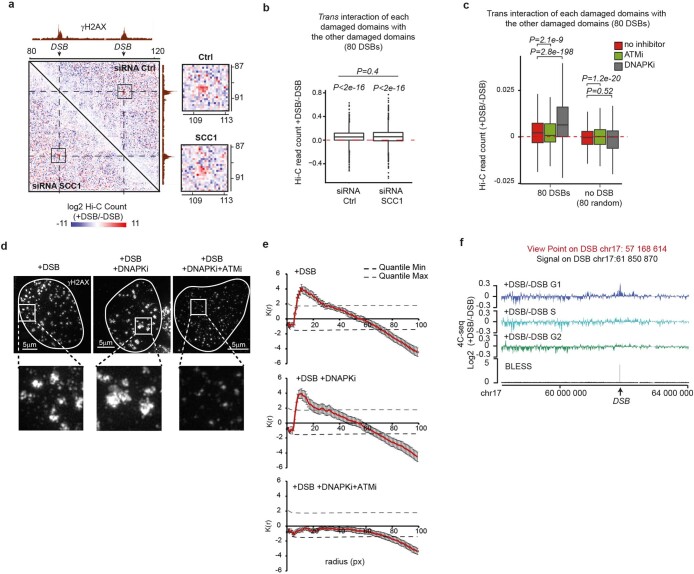
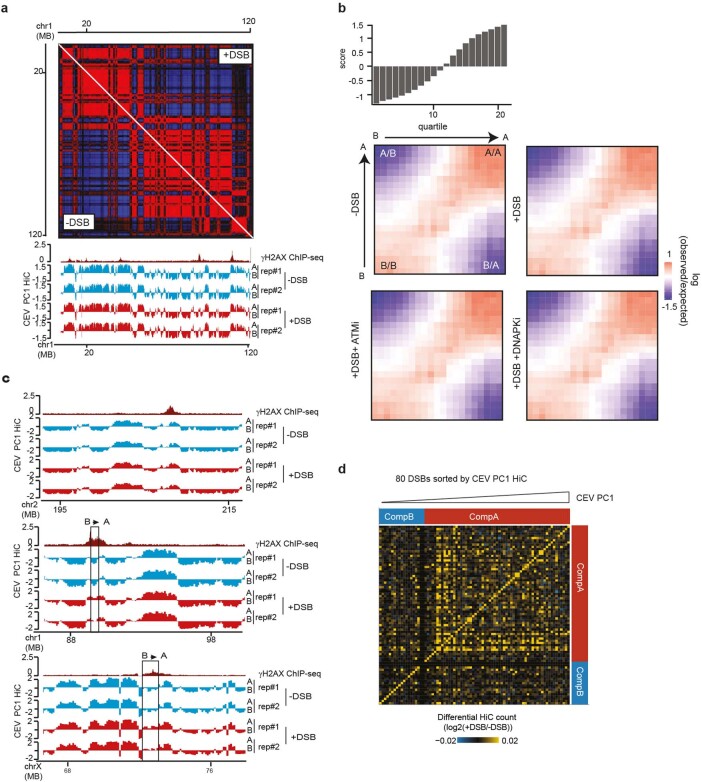
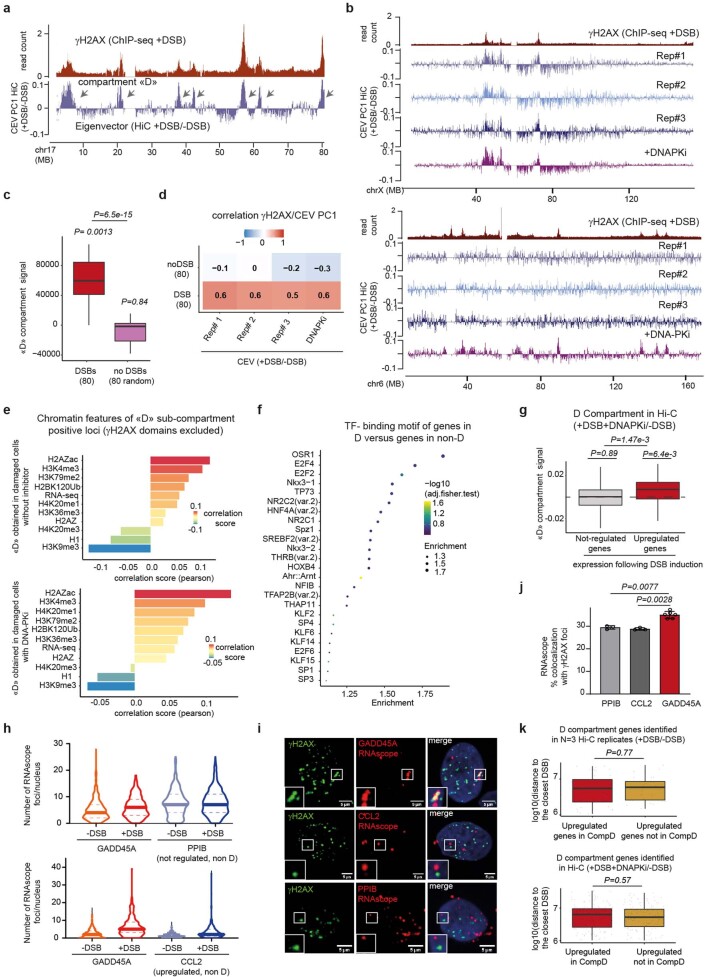
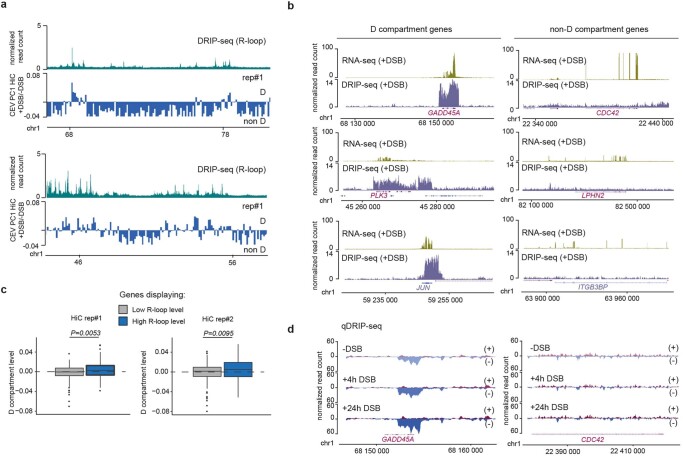
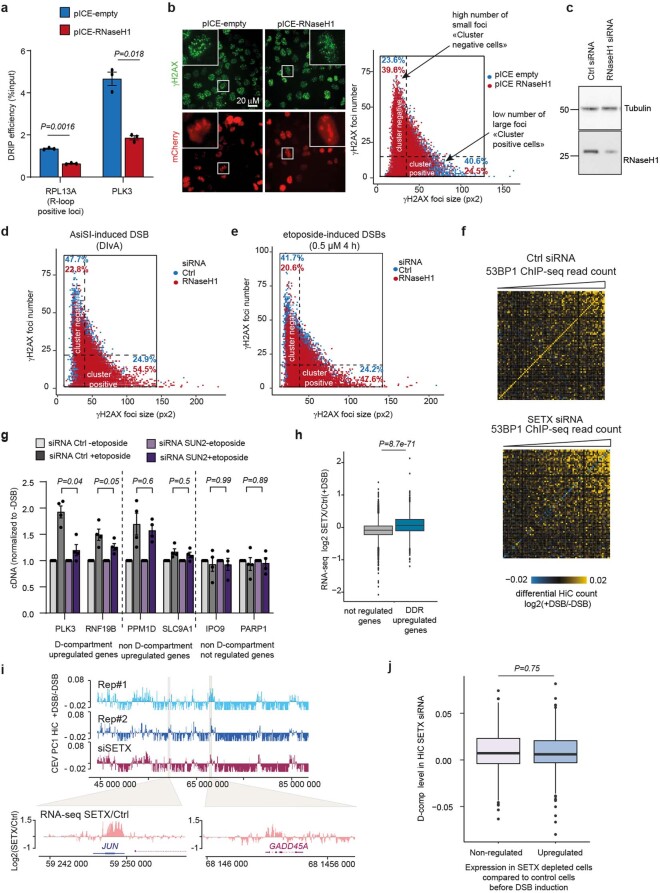
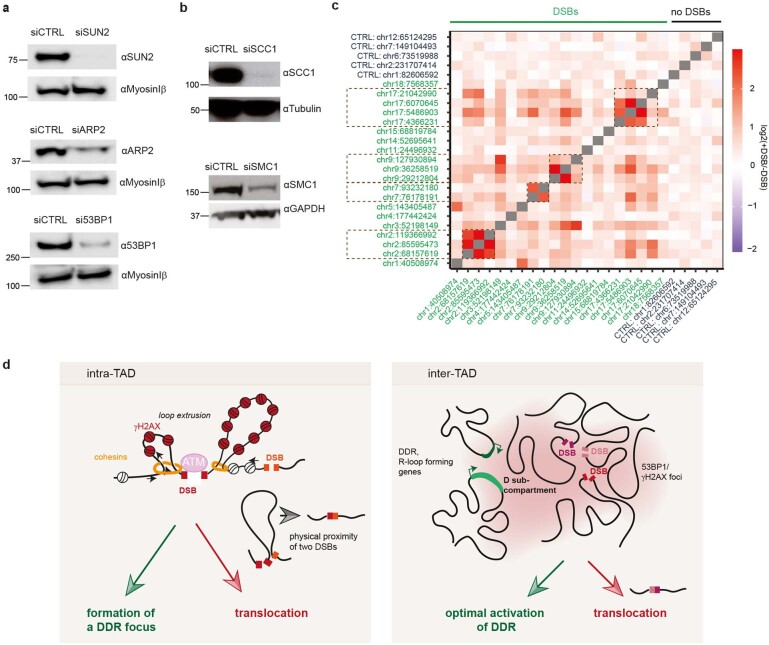
The DNA damage response is essential to safeguard genome integrity. Although the contribution of chromatin in DNA repair has been investigated1,2, the contribution of chromosome folding to these processes remains unclear3. Here we report that, after the production of double-stranded breaks (DSBs) in mammalian cells, ATM drives the formation of a new chromatin compartment (D compartment) through the clustering of damaged topologically associating domains, decorated with γH2AX and 53BP1. This compartment forms by a mechanism that is consistent with polymer-polymer phase separation rather than liquid-liquid phase separation. The D compartment arises mostly in G1 phase, is independent of cohesin and is enhanced after pharmacological inhibition of DNA-dependent protein kinase (DNA-PK) or R-loop accumulation. Importantly, R-loop-enriched DNA-damage-responsive genes physically localize to the D compartment, and this contributes to their optimal activation, providing a function for DSB clustering in the DNA damage response. However, DSB-induced chromosome reorganization comes at the expense of an increased rate of translocations, also observed in cancer genomes. Overall, we characterize how DSB-induced compartmentalization orchestrates the DNA damage response and highlight the critical impact of chromosome architecture in genomic instability.
© 2023. The Author(s).
Conflict of interest statement
N.A. is a cofounder and on the scientific advisory board of Regel Therapeutics and receives funding from BioMarin Pharmaceutical Inc. The other authors declare no competing interests.
Figures
Comment in
-
Walking the tightrope between DNA damage repair and genomic integrity.Neuro Oncol. 2024 Mar 4;26(3):399-400. doi: 10.1093/neuonc/noad218. Neuro Oncol. 2024. PMID: 37963038 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
