

Identification of the regulatory circuit governing corneal epithelial fate determination and disease
- PMID: 37856539
- PMCID: PMC10586658
- DOI: 10.1371/journal.pbio.3002336
Identification of the regulatory circuit governing corneal epithelial fate determination and disease
Abstract
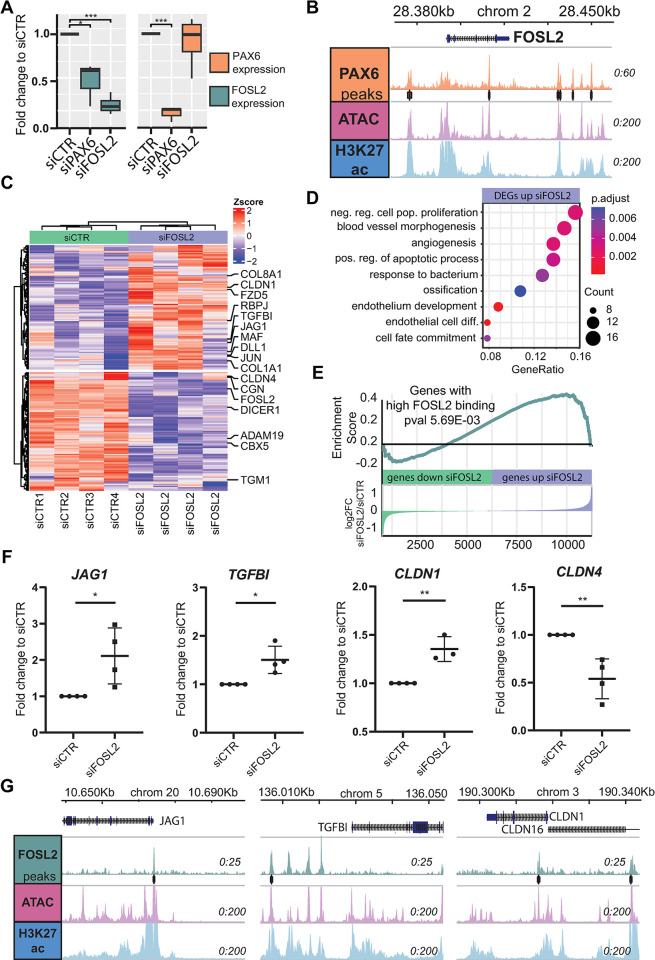
The transparent corneal epithelium in the eye is maintained through the homeostasis regulated by limbal stem cells (LSCs), while the nontransparent epidermis relies on epidermal keratinocytes for renewal. Despite their cellular similarities, the precise cell fates of these two types of epithelial stem cells, which give rise to functionally distinct epithelia, remain unknown. We performed a multi-omics analysis of human LSCs from the cornea and keratinocytes from the epidermis and characterized their molecular signatures, highlighting their similarities and differences. Through gene regulatory network analyses, we identified shared and cell type-specific transcription factors (TFs) that define specific cell fates and established their regulatory hierarchy. Single-cell RNA-seq (scRNA-seq) analyses of the cornea and the epidermis confirmed these shared and cell type-specific TFs. Notably, the shared and LSC-specific TFs can cooperatively target genes associated with corneal opacity. Importantly, we discovered that FOSL2, a direct PAX6 target gene, is a novel candidate associated with corneal opacity, and it regulates genes implicated in corneal diseases. By characterizing molecular signatures, our study unveils the regulatory circuitry governing the LSC fate and its association with corneal opacity.
Copyright: © 2023 Smits et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
