Evolution of haploid and diploid populations reveals common, strong, and variable pleiotropic effects in non-home environments
- PMID: 37861305
- PMCID: PMC10629826
- DOI: 10.7554/eLife.92899
Evolution of haploid and diploid populations reveals common, strong, and variable pleiotropic effects in non-home environments
Abstract
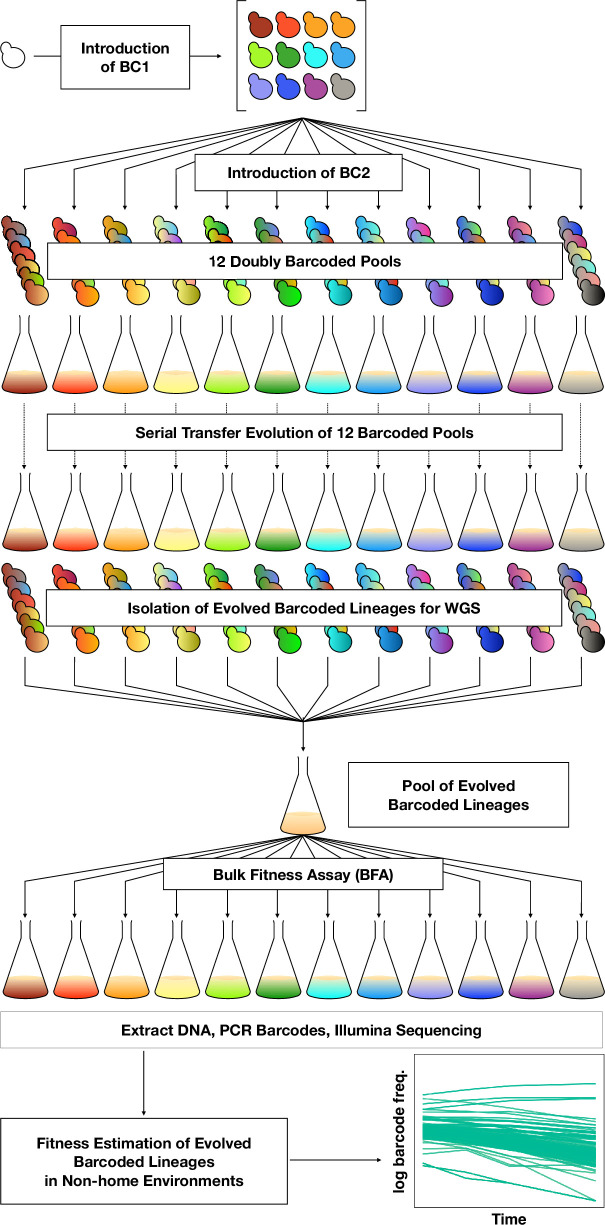
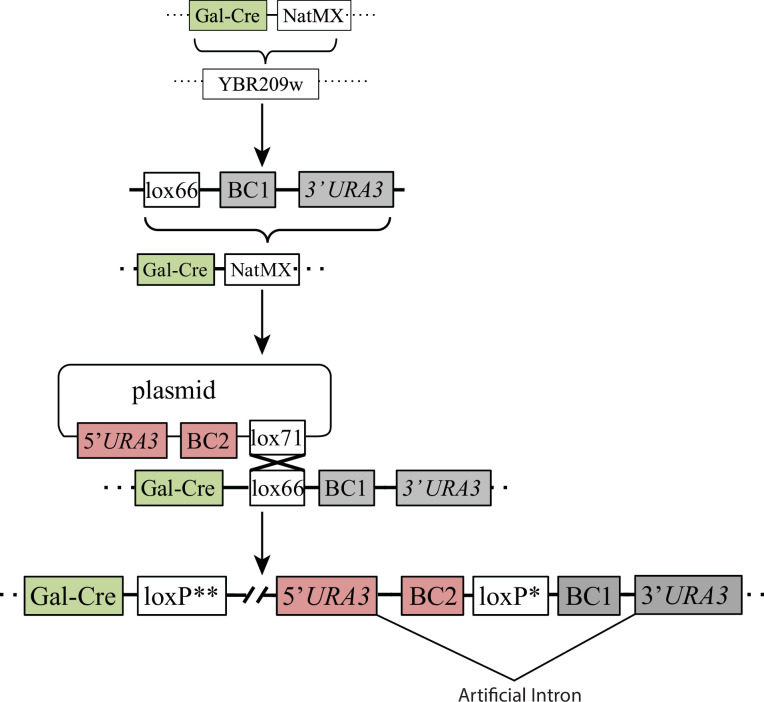
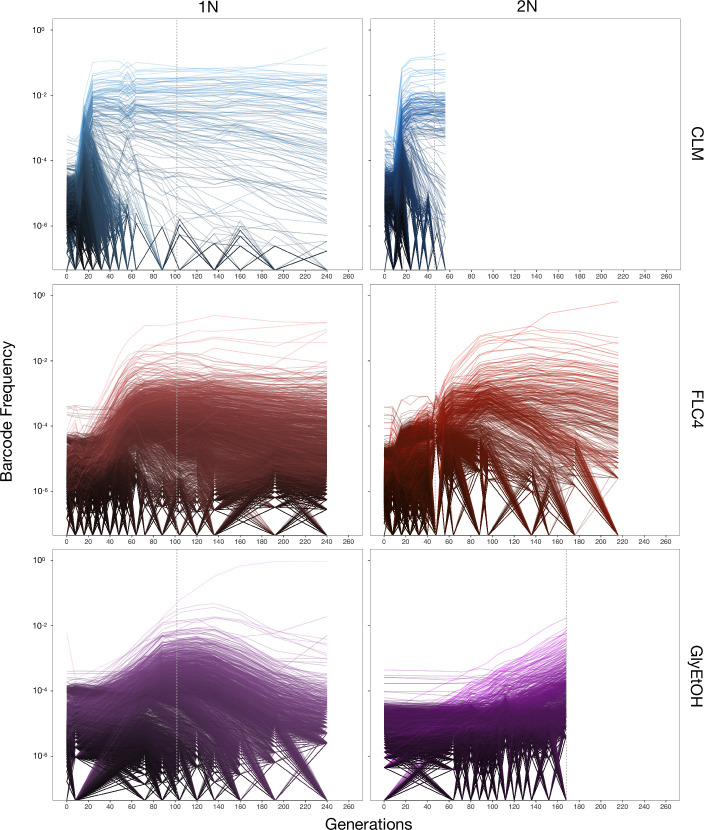
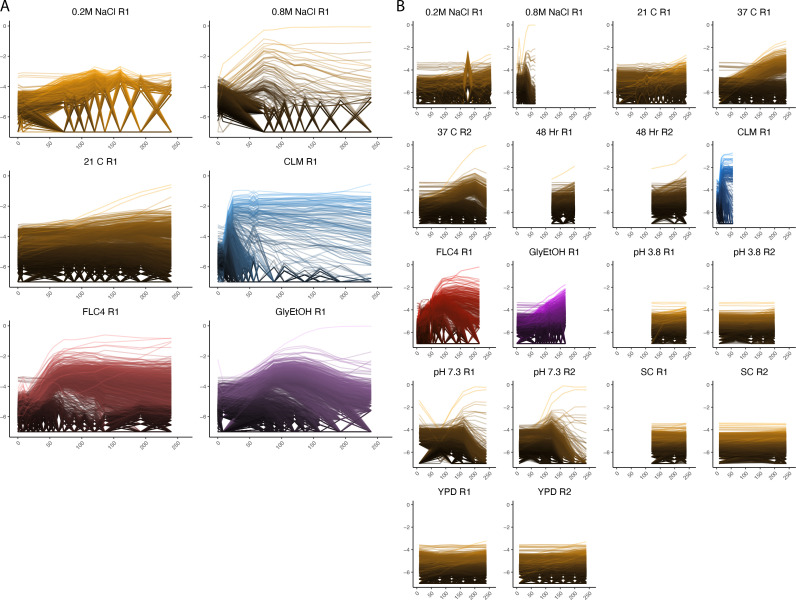
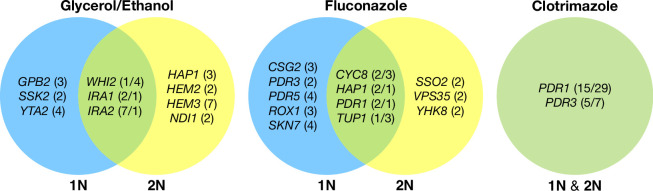
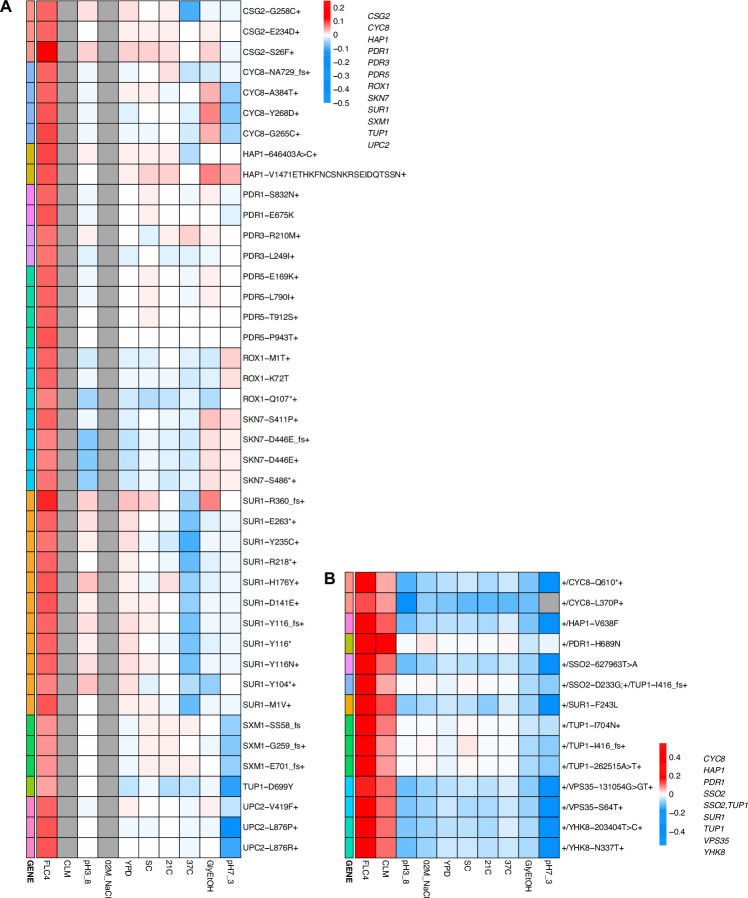
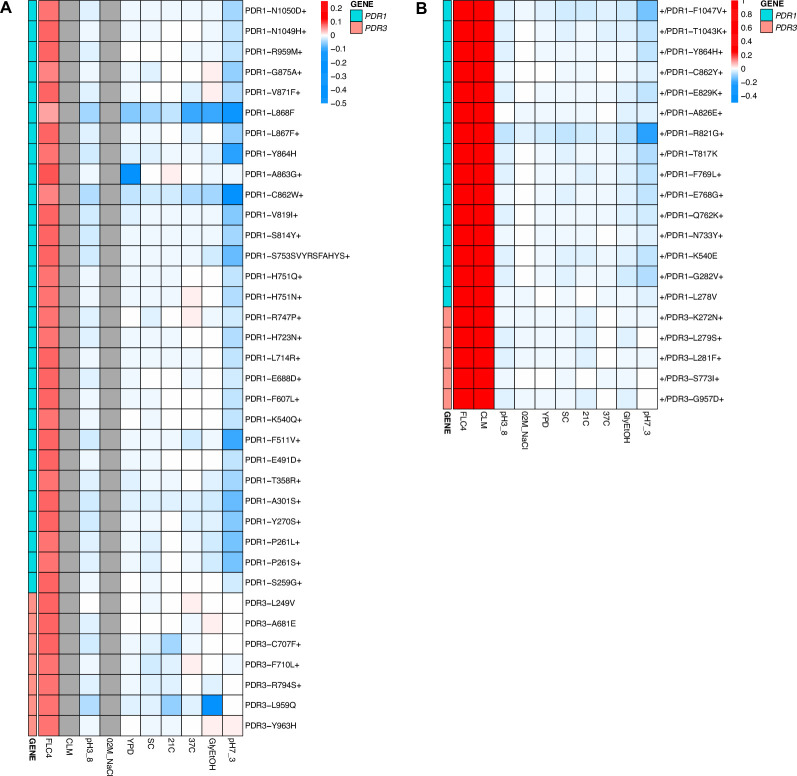
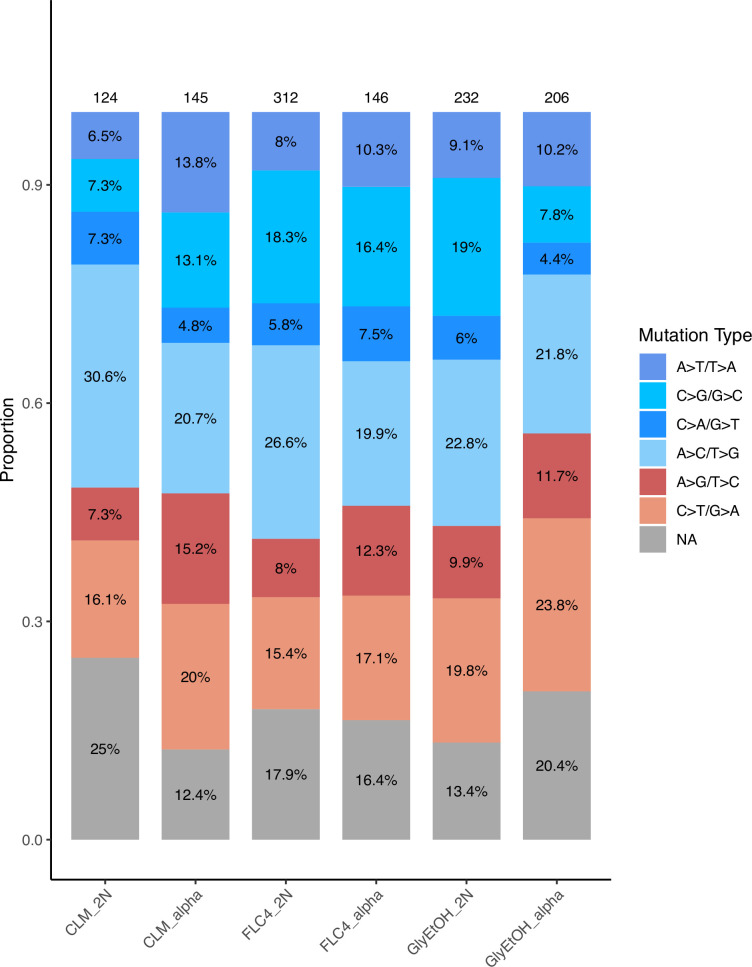
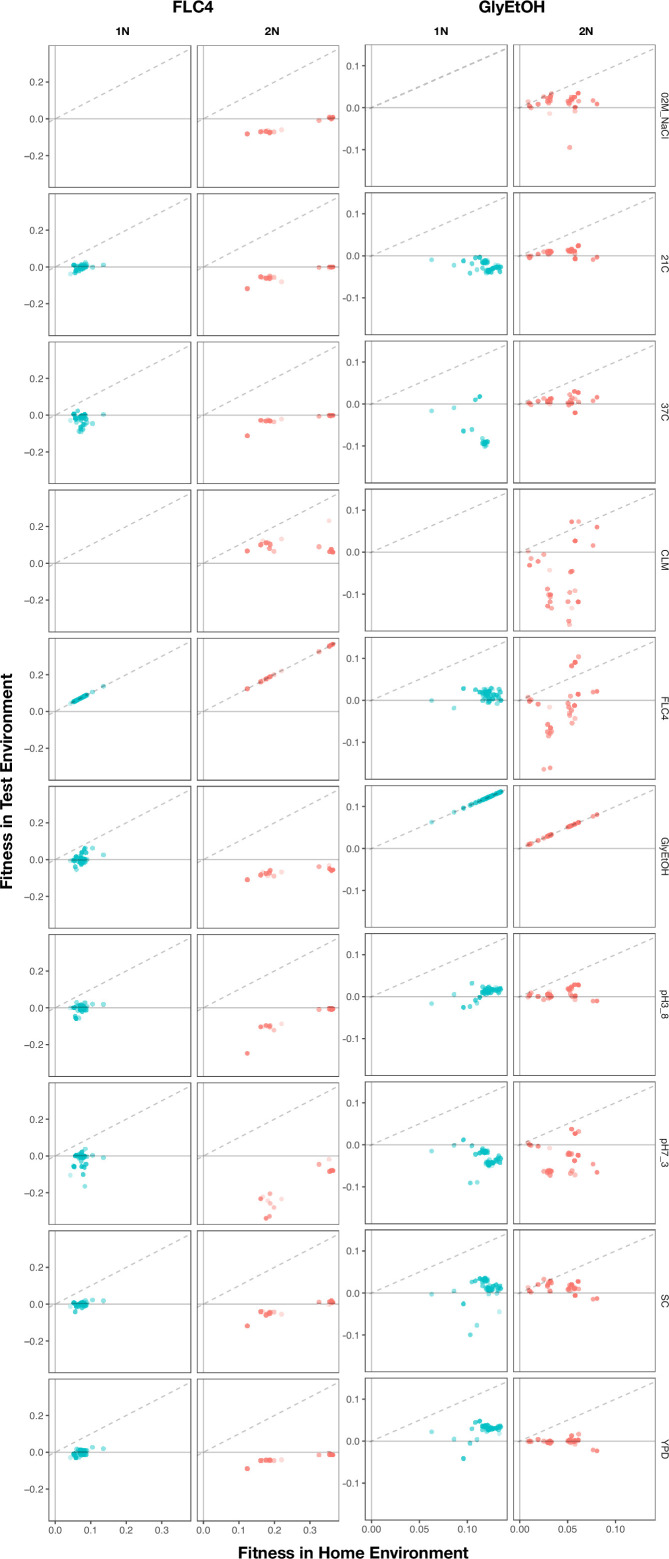
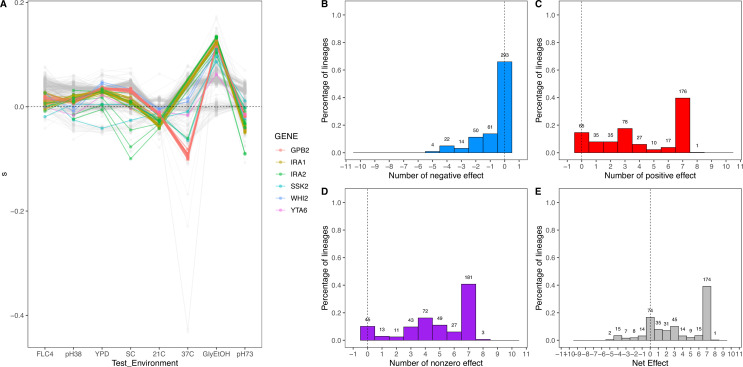
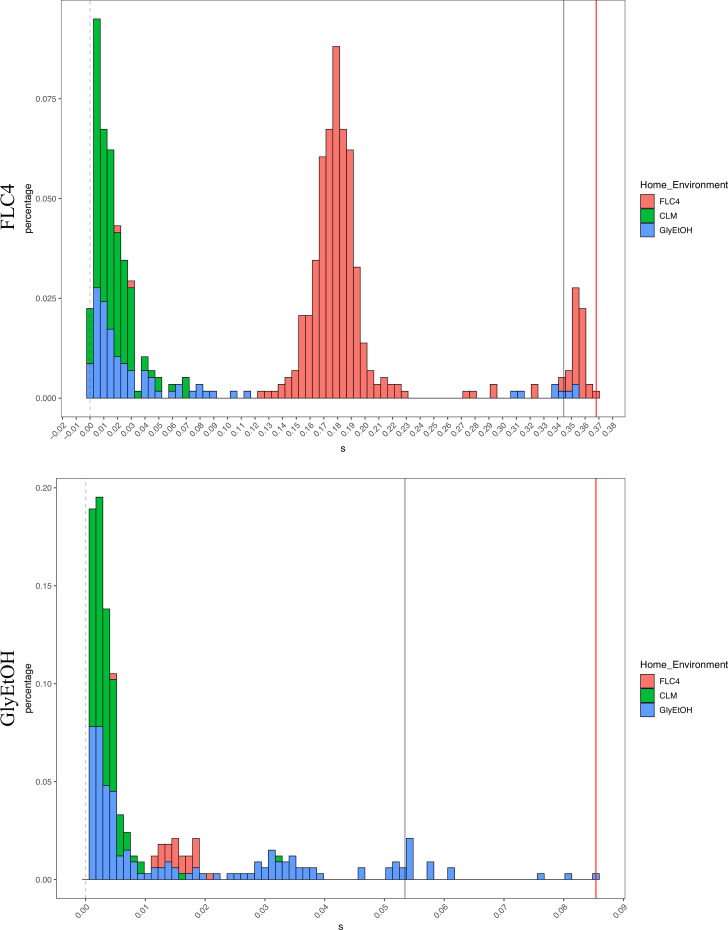
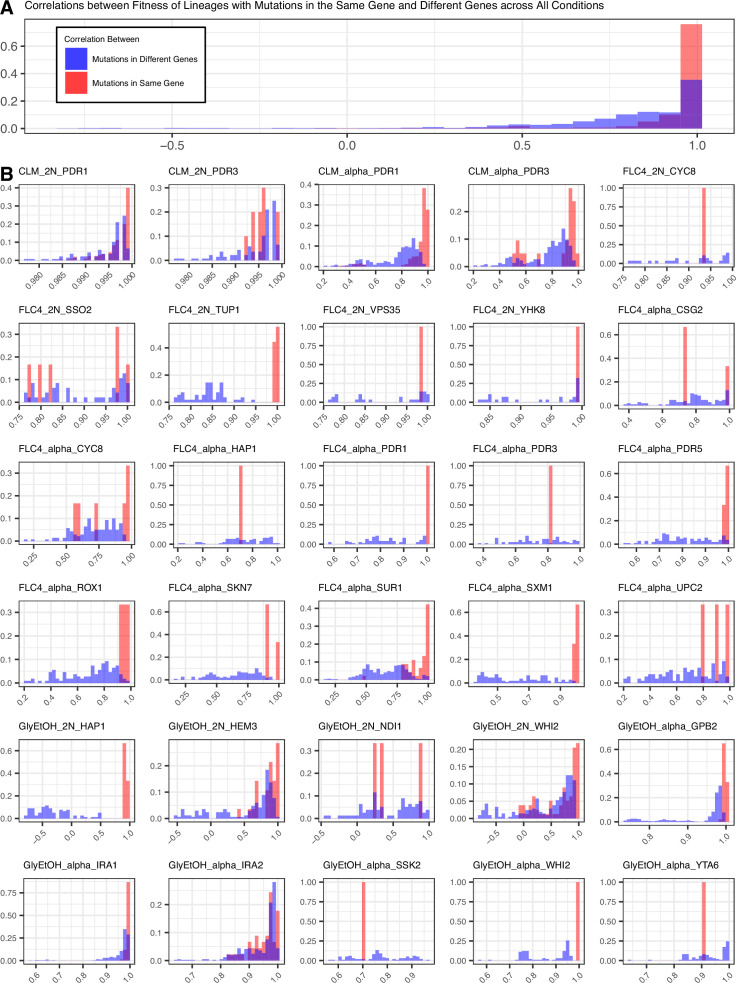
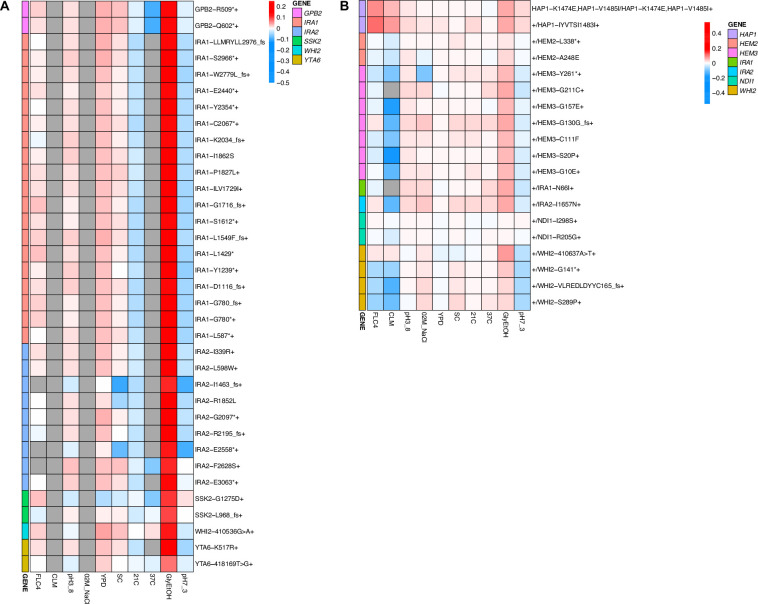
Adaptation is driven by the selection for beneficial mutations that provide a fitness advantage in the specific environment in which a population is evolving. However, environments are rarely constant or predictable. When an organism well adapted to one environment finds itself in another, pleiotropic effects of mutations that made it well adapted to its former environment will affect its success. To better understand such pleiotropic effects, we evolved both haploid and diploid barcoded budding yeast populations in multiple environments, isolated adaptive clones, and then determined the fitness effects of adaptive mutations in 'non-home' environments in which they were not selected. We find that pleiotropy is common, with most adaptive evolved lineages showing fitness effects in non-home environments. Consistent with other studies, we find that these pleiotropic effects are unpredictable: they are beneficial in some environments and deleterious in others. However, we do find that lineages with adaptive mutations in the same genes tend to show similar pleiotropic effects. We also find that ploidy influences the observed adaptive mutational spectra in a condition-specific fashion. In some conditions, haploids and diploids are selected with adaptive mutations in identical genes, while in others they accumulate mutations in almost completely disjoint sets of genes.
Keywords: S. cerevisiae; evolution; evolutionary biology; genetics; genomics; pleiotropy; trade-offs; yeast.
© 2023, Chen, Johnson, Hérissant et al.
Conflict of interest statement
VC, MJ, LH, PH, DY, YL, AA, SH, DP, MD, GS No competing interests declared
Figures