

Imaging and multi-omics datasets converge to define different neural progenitor origins for ATRT-SHH subgroups
- PMID: 37863903
- PMCID: PMC10589300
- DOI: 10.1038/s41467-023-42371-7
Imaging and multi-omics datasets converge to define different neural progenitor origins for ATRT-SHH subgroups
Abstract
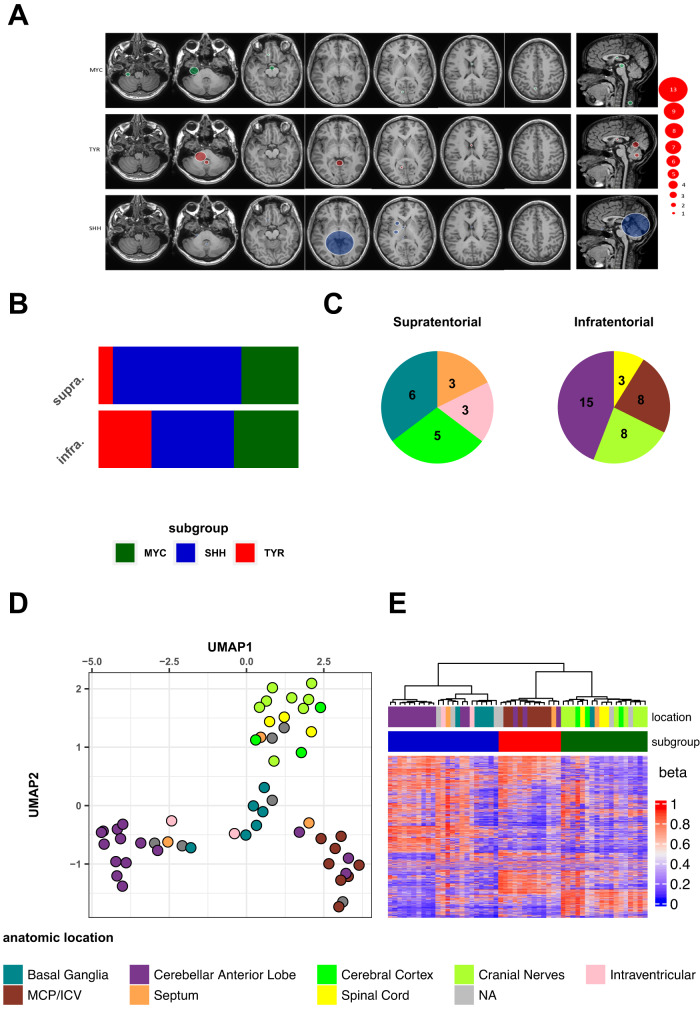
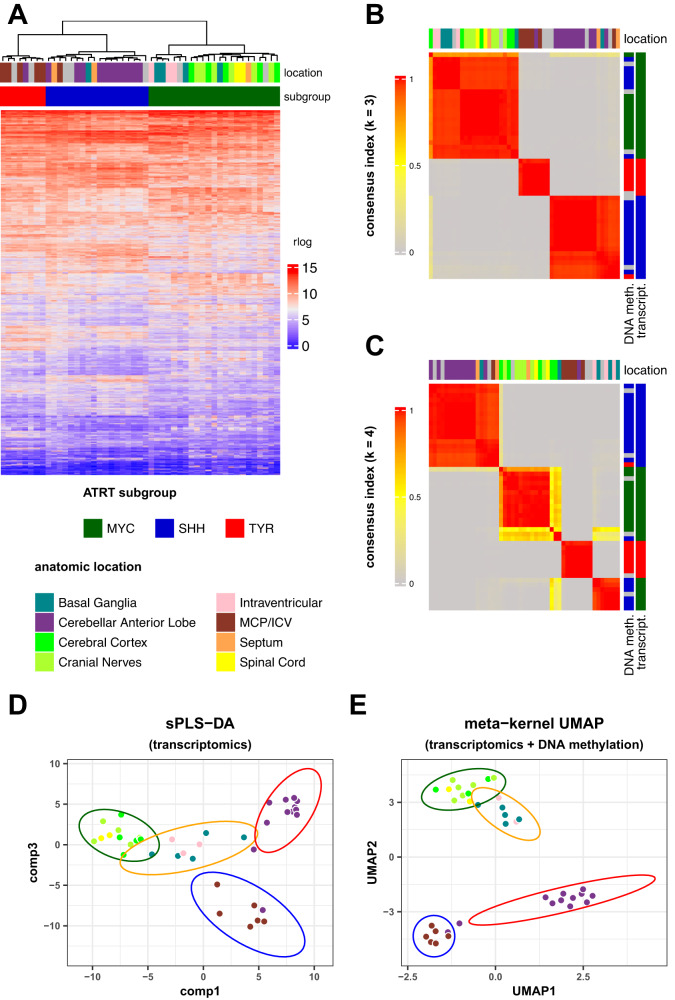
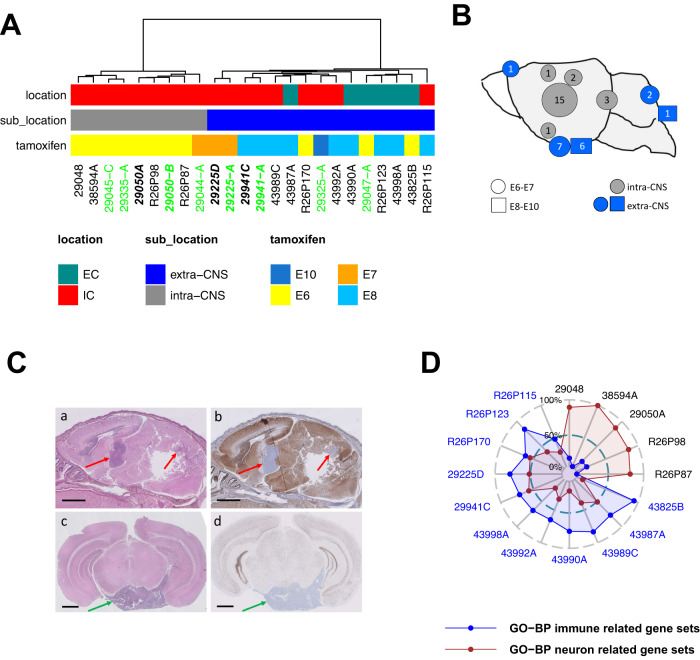
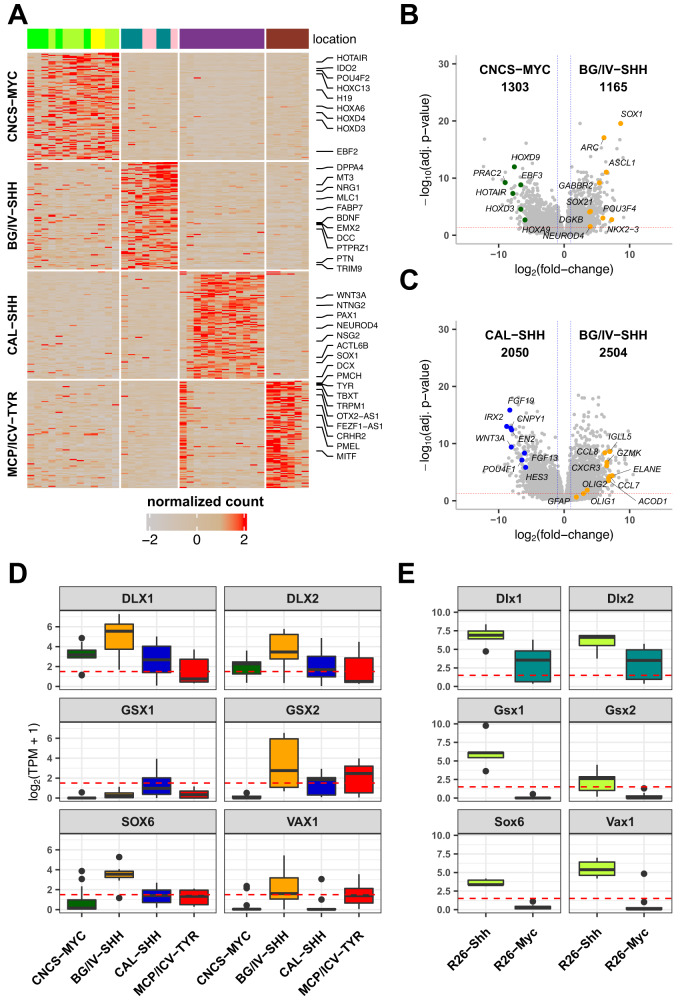
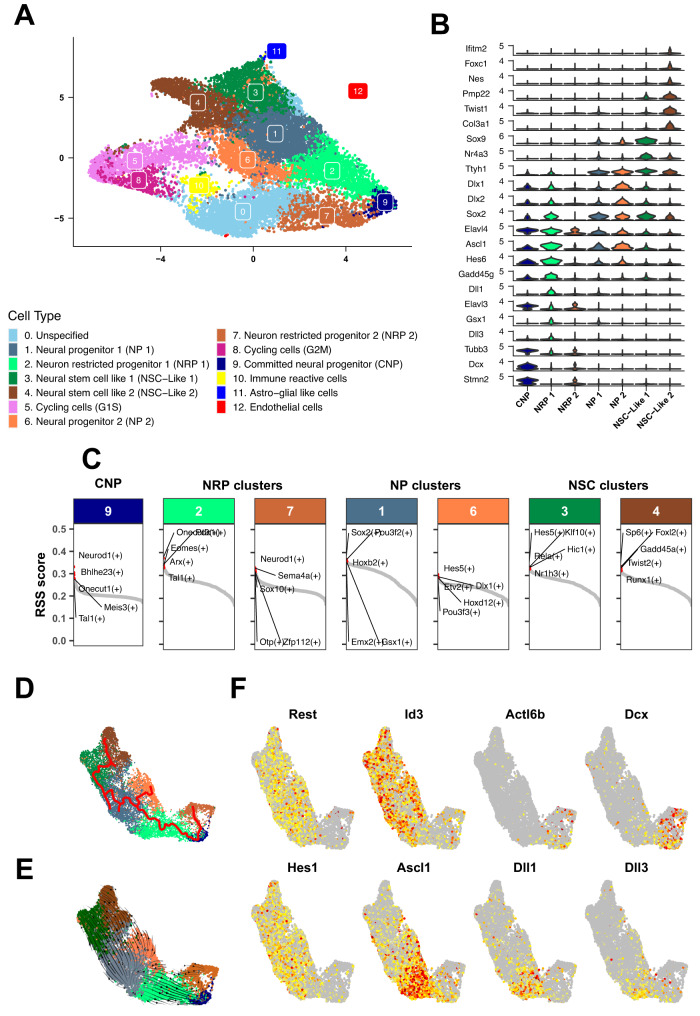
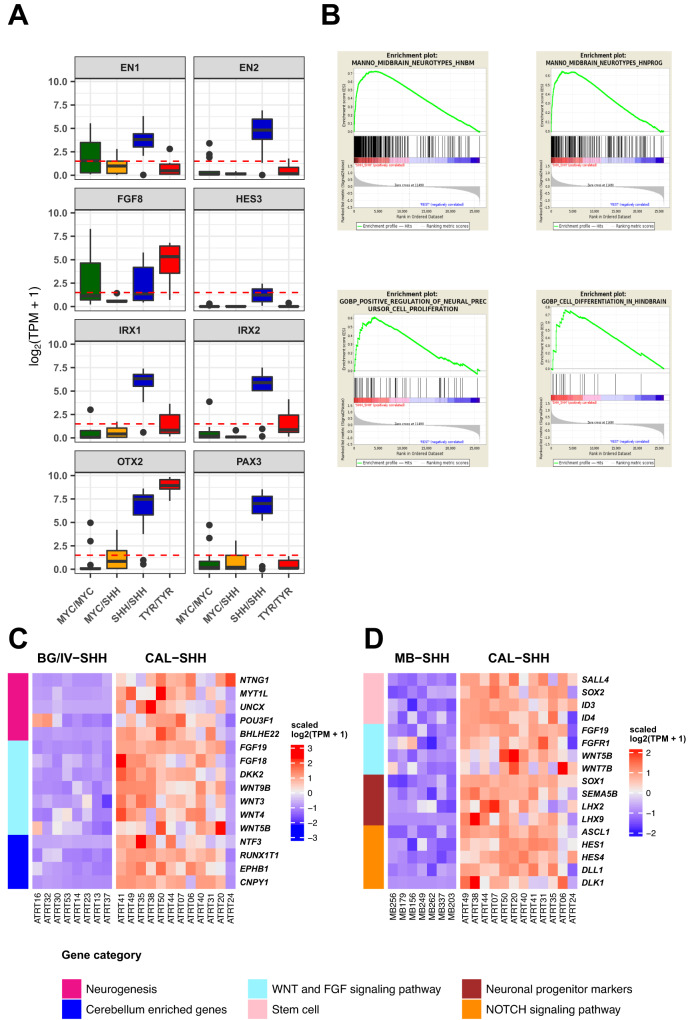
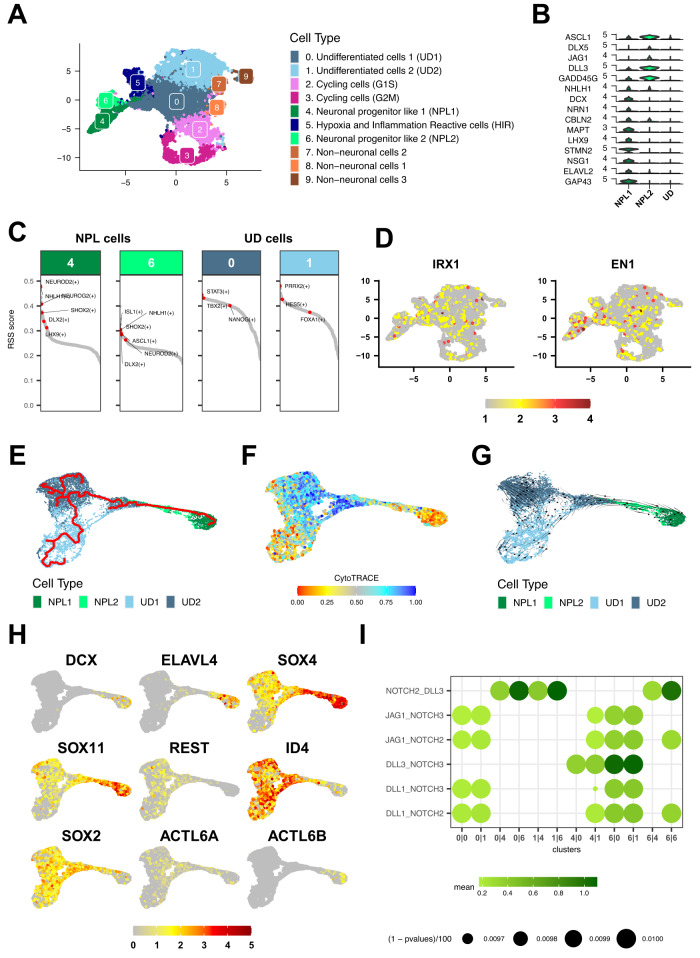
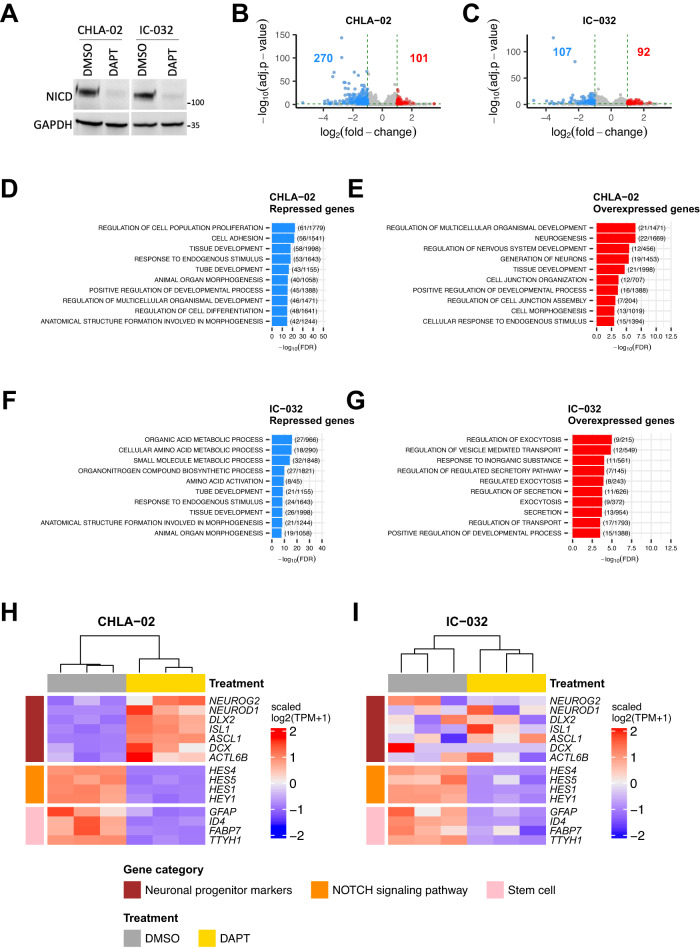
Atypical teratoid rhabdoid tumors (ATRT) are divided into MYC, TYR and SHH subgroups, suggesting diverse lineages of origin. Here, we investigate the imaging of human ATRT at diagnosis and the precise anatomic origin of brain tumors in the Rosa26-CreERT2::Smarcb1flox/flox model. This cross-species analysis points to an extra-cerebral origin for MYC tumors. Additionally, we clearly distinguish SHH ATRT emerging from the cerebellar anterior lobe (CAL) from those emerging from the basal ganglia (BG) and intra-ventricular (IV) regions. Molecular characteristics point to the midbrain-hindbrain boundary as the origin of CAL SHH ATRT, and to the ganglionic eminence as the origin of BG/IV SHH ATRT. Single-cell RNA sequencing on SHH ATRT supports these hypotheses. Trajectory analyses suggest that SMARCB1 loss induces a de-differentiation process mediated by repressors of the neuronal program such as REST, ID and the NOTCH pathway.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Johann PD, et al. Atypical Teratoid/Rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29:379–393. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
