

Identification of a robust DNA methylation signature for Fanconi anemia
- PMID: 37865086
- PMCID: PMC10645556
- DOI: 10.1016/j.ajhg.2023.09.014
Identification of a robust DNA methylation signature for Fanconi anemia
Abstract
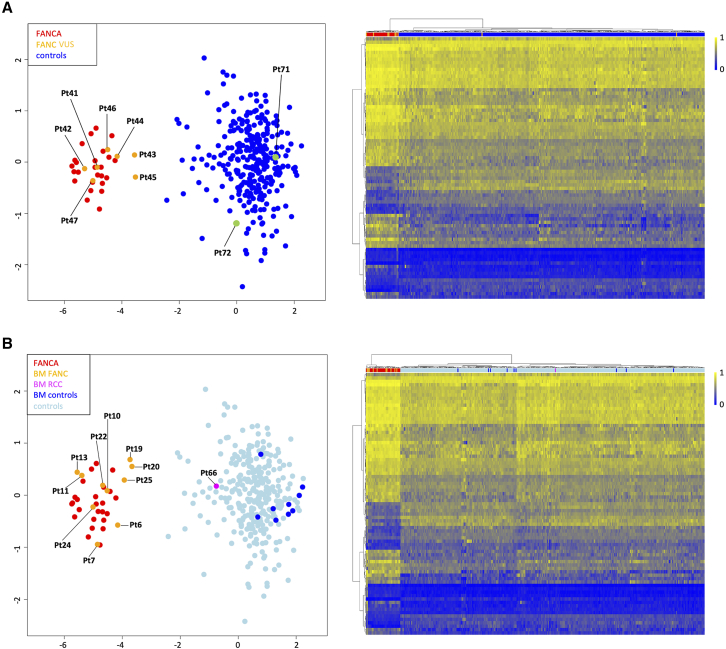
Fanconi anemia (FA) is a clinically variable and genetically heterogeneous cancer-predisposing disorder representing the most common bone marrow failure syndrome. It is caused by inactivating predominantly biallelic mutations involving >20 genes encoding proteins with roles in the FA/BRCA DNA repair pathway. Molecular diagnosis of FA is challenging due to the wide spectrum of the contributing gene mutations and structural rearrangements. The assessment of chromosomal fragility after exposure to DNA cross-linking agents is generally required to definitively confirm diagnosis. We assessed peripheral blood genome-wide DNA methylation (DNAm) profiles in 25 subjects with molecularly confirmed clinical diagnosis of FA (FANCA complementation group) using Illumina's Infinium EPIC array. We identified 82 differentially methylated CpG sites that allow to distinguish subjects with FA from healthy individuals and subjects with other genetic disorders, defining an FA-specific DNAm signature. The episignature was validated using a second cohort of subjects with FA involving different complementation groups, documenting broader genetic sensitivity and demonstrating its specificity using the EpiSign Knowledge Database. The episignature properly classified DNA samples obtained from bone marrow aspirates, demonstrating robustness. Using the selected probes, we trained a machine-learning model able to classify EPIC DNAm profiles in molecularly unsolved cases. Finally, we show that the generated episignature includes CpG sites that do not undergo functional selective pressure, allowing diagnosis of FA in individuals with reverted phenotype due to gene conversion. These findings provide a tool to accelerate diagnostic testing in FA and broaden the clinical utility of DNAm profiling in the diagnostic setting.
Keywords: DNA methylation profiling; Fanconi anemia; classifier; diagnostic tool; episignature; gene conversion; hematological disorders; machine learning; mosaicism; variant classification.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Dr. Sadikovic is a shareholder in EpiSign Inc, a software company involved in commercialization of EpiSign Technology.
Figures




References
-
- Kutler D.I., Singh B., Satagopan J., Batish S.D., Berwick M., Giampietro P.F., Hanenberg H., Auerbach A.D. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101:1249–1256. - PubMed
-
- Mehta P.A., Ebens C. In: GeneReviews Seattle (WA) Adam M.P., Mirzaa G.M., Pagon R.A., Wallace S.E., Bean L.J., Gripp K.W., Amemiya A., editors. University of Washington, Seattle; 2002. Fanconi Anemia; pp. 1993–2023.
-
- Schneider M., Chandler K., Tischkowitz M., Meyer S. Fanconi anemia: genetics, molecular biology, and cancer – implications for clinical management in children and adults. Clin. Genet. 2015;88:13–24. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
