

Liquid biopsy epigenomic profiling for cancer subtyping
- PMID: 37865722
- PMCID: PMC10695830
- DOI: 10.1038/s41591-023-02605-z
Liquid biopsy epigenomic profiling for cancer subtyping
Erratum in
-
Author Correction: Liquid biopsy epigenomic profiling for cancer subtyping.Nat Med. 2024 Mar;30(3):907. doi: 10.1038/s41591-023-02735-4. Nat Med. 2024. PMID: 38049623 Free PMC article. No abstract available.
Abstract
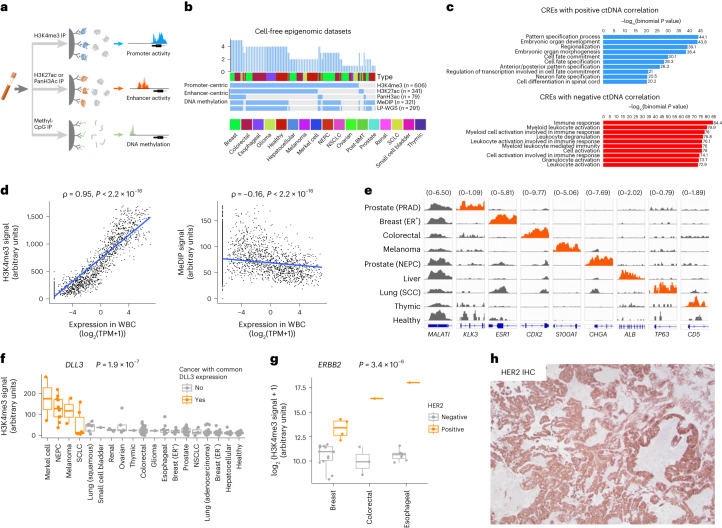
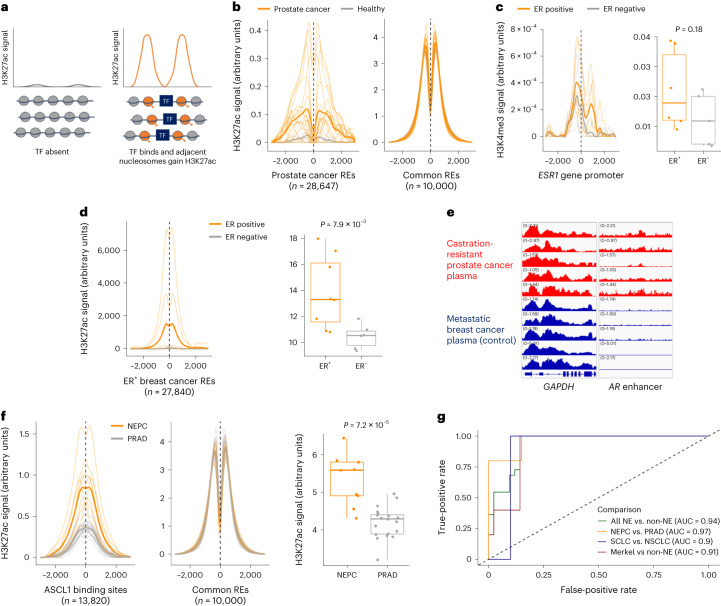
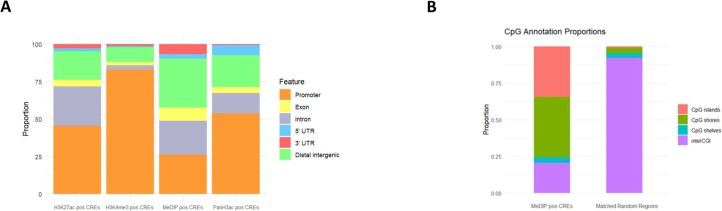
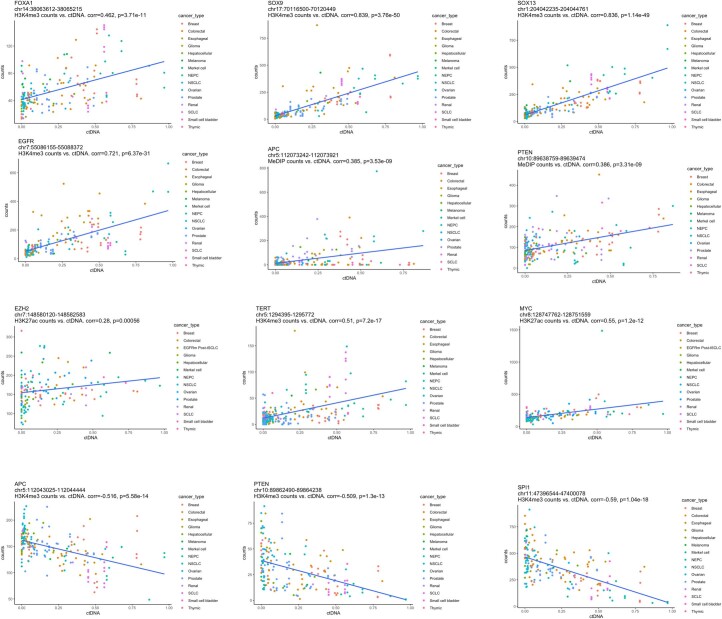
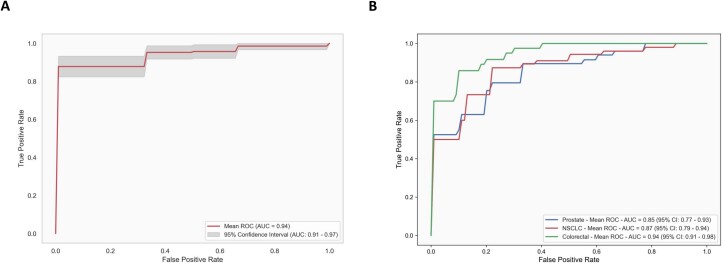
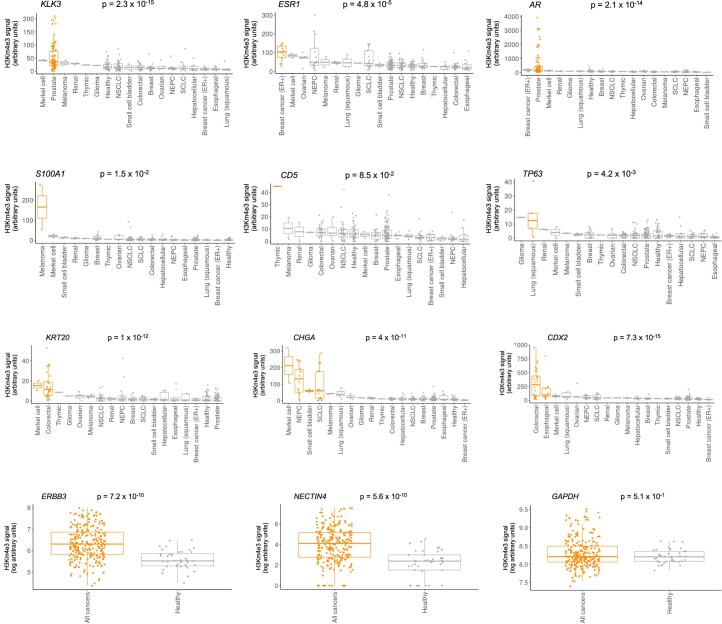
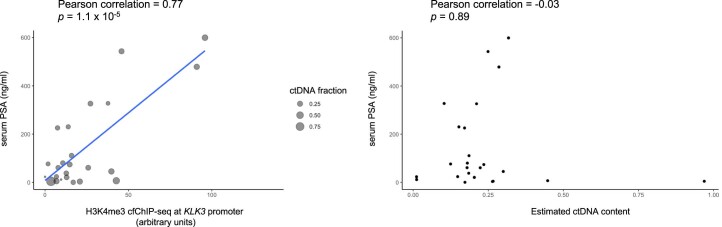
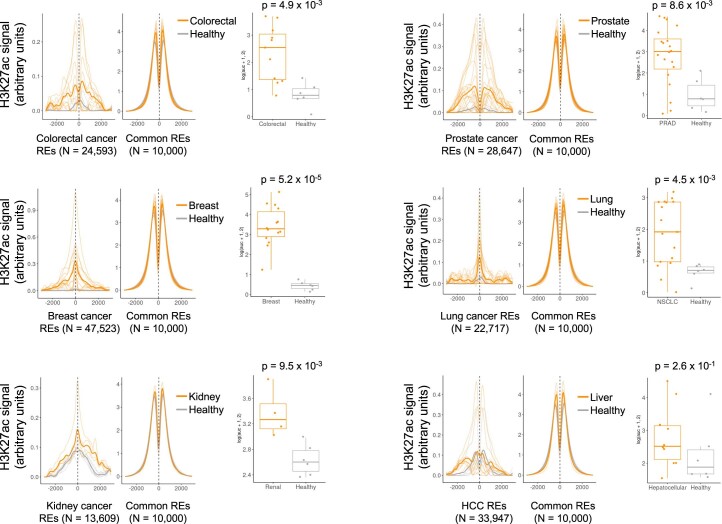
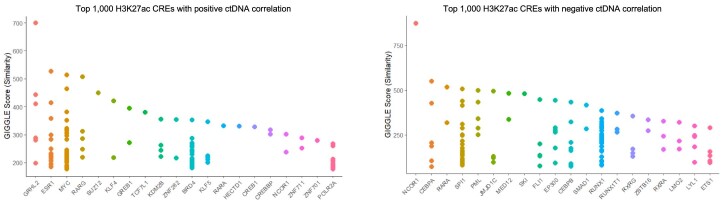
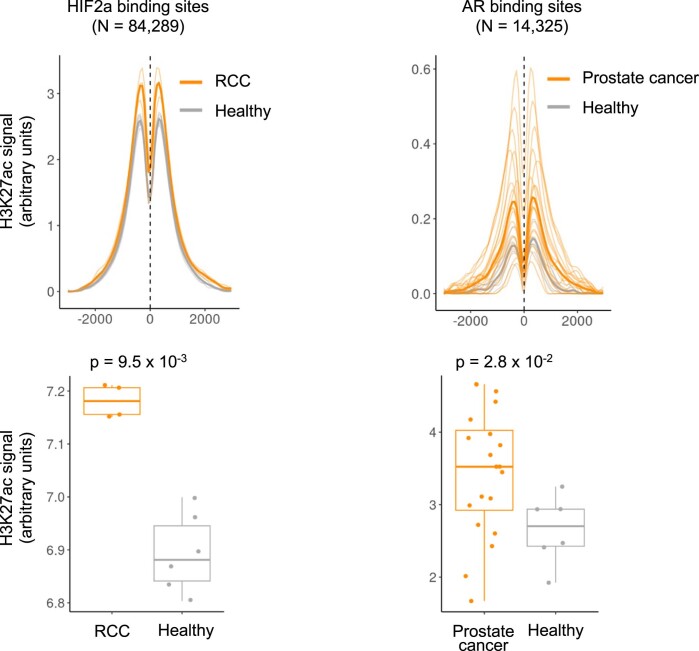
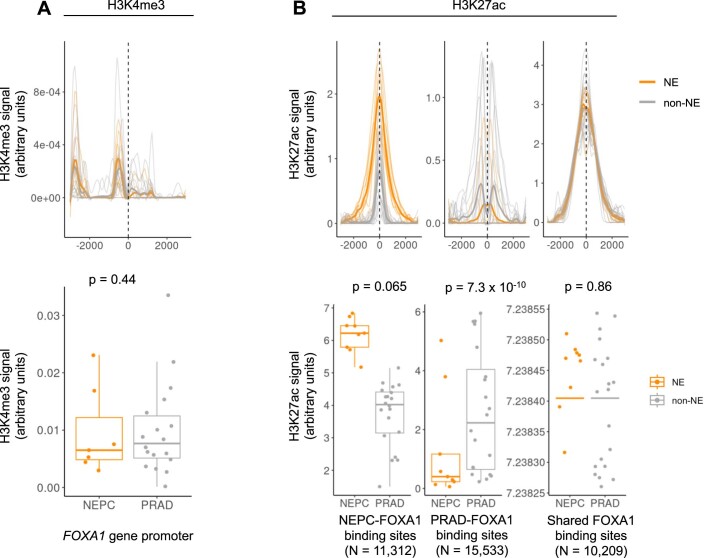
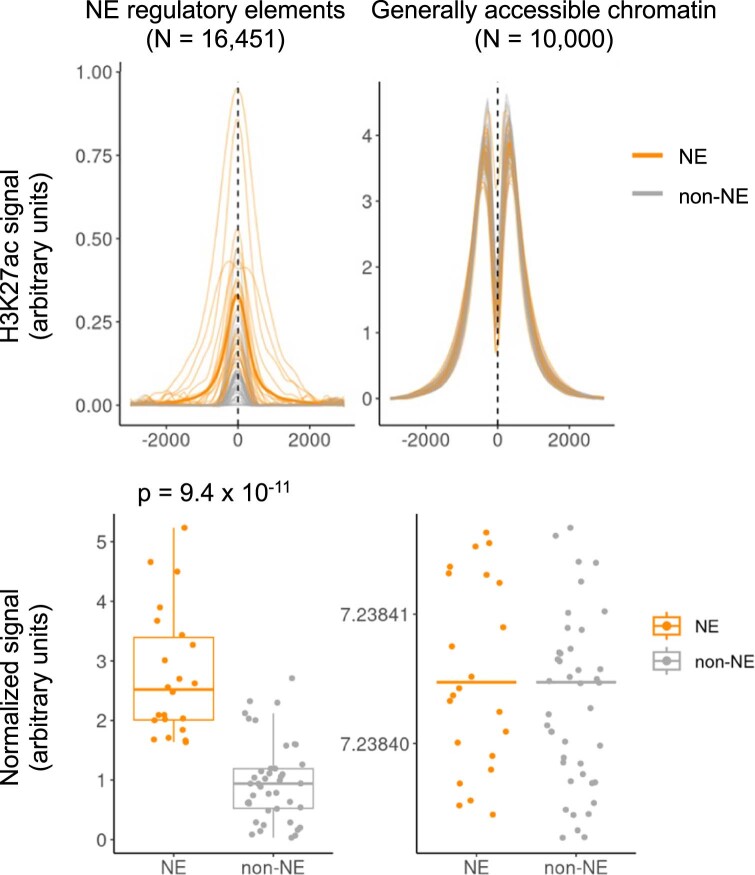
Although circulating tumor DNA (ctDNA) assays are increasingly used to inform clinical decisions in cancer care, they have limited ability to identify the transcriptional programs that govern cancer phenotypes and their dynamic changes during the course of disease. To address these limitations, we developed a method for comprehensive epigenomic profiling of cancer from 1 ml of patient plasma. Using an immunoprecipitation-based approach targeting histone modifications and DNA methylation, we measured 1,268 epigenomic profiles in plasma from 433 individuals with one of 15 cancers. Our assay provided a robust proxy for transcriptional activity, allowing us to infer the expression levels of diagnostic markers and drug targets, measure the activity of therapeutically targetable transcription factors and detect epigenetic mechanisms of resistance. This proof-of-concept study in advanced cancers shows how plasma epigenomic profiling has the potential to unlock clinically actionable information that is currently accessible only via direct tissue sampling.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
S.C.B., T.K.C. and M.L.F. are co-founders and shareholders of Precede Biosciences. J.D. is a consultant for Kymera Therapeutics and has a sponsored research agreement with Kymera Therapeutics. M.T. served on an advisory board for Incyte. A.N.H. reports research support from Amgen, Blueprint Medicines, BridgeBio, Bristol-Myers Squibb, C4 Therapeutics, Eli Lilly, Novartis, Nuvalent, Pfizer, Roche/Genentech and Scorpion Therapeutics and paid consulting for Engine Biosciences, Nuvalent, Oncovalent, TigaTx and Tolremo Therapeutics. J.R. receives research funding from Equillium, Kite/Gilead, Novartis and Oncternal and consults or is on advisory boards for AvroBio, Akron Biotech, Clade Therapeutics, Garuda Therapeutics, LifeVault Bio, Novartis, Smart Immune and TScan Therapeutics. The remaining authors report no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
