

Metagenomic Insight into The Global Dissemination of The Antibiotic Resistome
- PMID: 37870180
- PMCID: PMC10667823
- DOI: 10.1002/advs.202303925
Metagenomic Insight into The Global Dissemination of The Antibiotic Resistome
Abstract
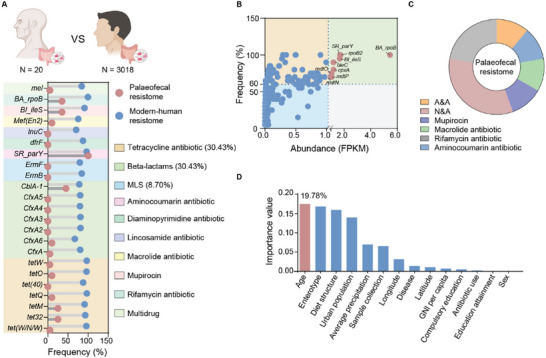
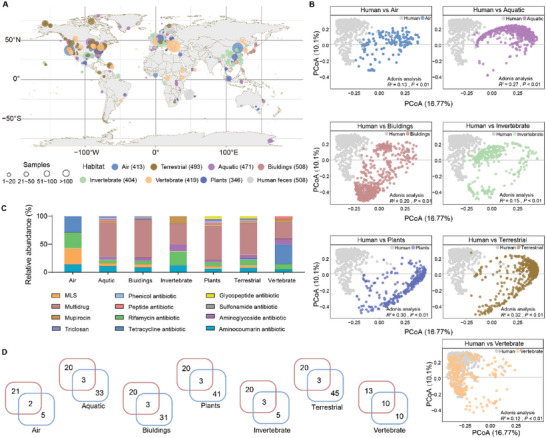
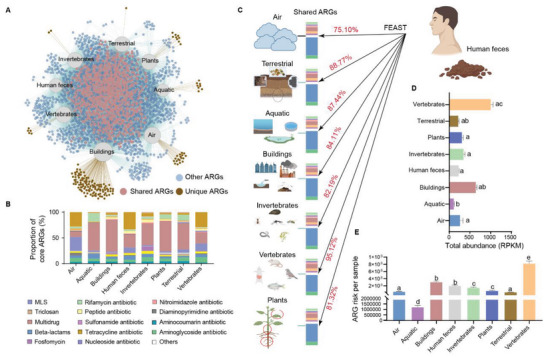
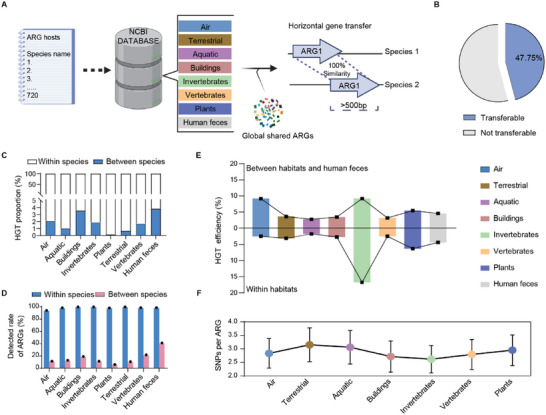
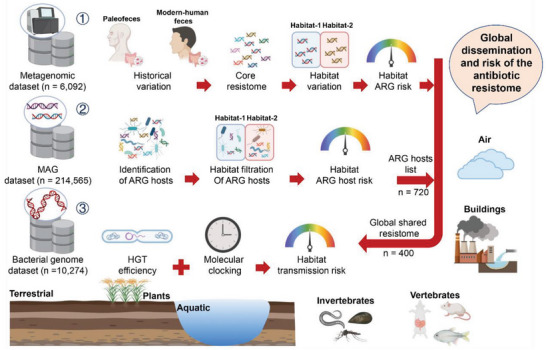
The global crisis in antimicrobial resistance continues to grow. Estimating the risks of antibiotic resistance transmission across habitats is hindered by the lack of data on mobility and habitat-specificity. Metagenomic samples of 6092 are analyzed to delineate the unique core resistomes from human feces and seven other habitats. This is found that most resistance genes (≈85%) are transmitted between external habitats and human feces. This suggests that human feces are broadly representative of the global resistome and are potentially a hub for accumulating and disseminating resistance genes. The analysis found that resistance genes with ancient horizontal gene transfer (HGT) events have a higher efficiency of transfer across habitats, suggesting that HGT may be the main driver for forming unique but partly shared resistomes in all habitats. Importantly, the human fecal resistome is historically different and influenced by HGT and age. The most important routes of cross-transmission of resistance are from the atmosphere, buildings, and animals to humans. These habitats should receive more attention for future prevention of antimicrobial resistance. The study will disentangle transmission routes of resistance genes between humans and other habitats in a One Health framework and can identify strategies for controlling the ongoing dissemination and antibiotic resistance.
Keywords: antibiotic resistome; horizontal gene transfer; metadata; metagenome; one health.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Afshin A., Sur P. J., Fay K. A., Cornaby L., Ferrara G., Salama J. S., Mullany E. C., Abate K. H., Abbafati C., Abebe Z., Afarideh M., Aggarwal A., Agrawal S., Akinyemiju T., Alahdab F., Bacha U., Bachman V. F., Badali H., Badawi A., Bensenor I. M., Bernabe E., Biadgilign S. K. K., Biryukov S. H., Cahill L. E., Carrero J. J., Cercy K. M., Dandona L., Dandona R., Dang A. K., Degefa M. G., et al., The Lancet 2019, 393, 1958.
-
- Programme, U. N. E., Bracing for Superbugs: Strengthening environmental action in the One Health response to antimicrobial resistance, https://www.unep.org/resources/superbugs/environmental‐action, Geneva, (Feb. 2023).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical