

Dual targeted extracellular vesicles regulate oncogenic genes in advanced pancreatic cancer
- PMID: 37872156
- PMCID: PMC10593751
- DOI: 10.1038/s41467-023-42402-3
Dual targeted extracellular vesicles regulate oncogenic genes in advanced pancreatic cancer
Abstract
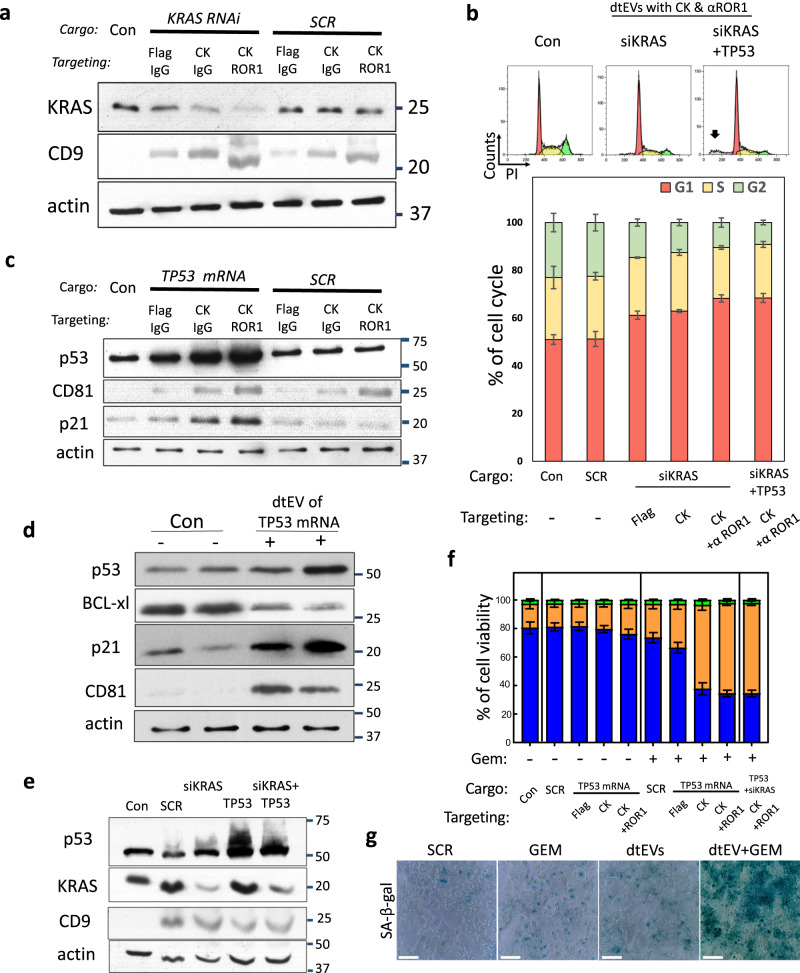
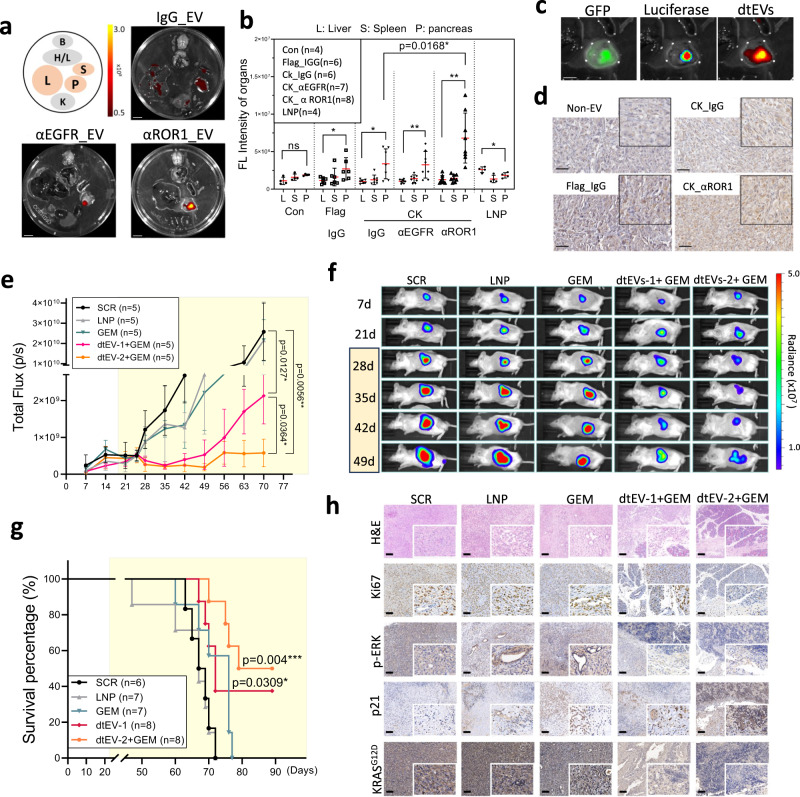
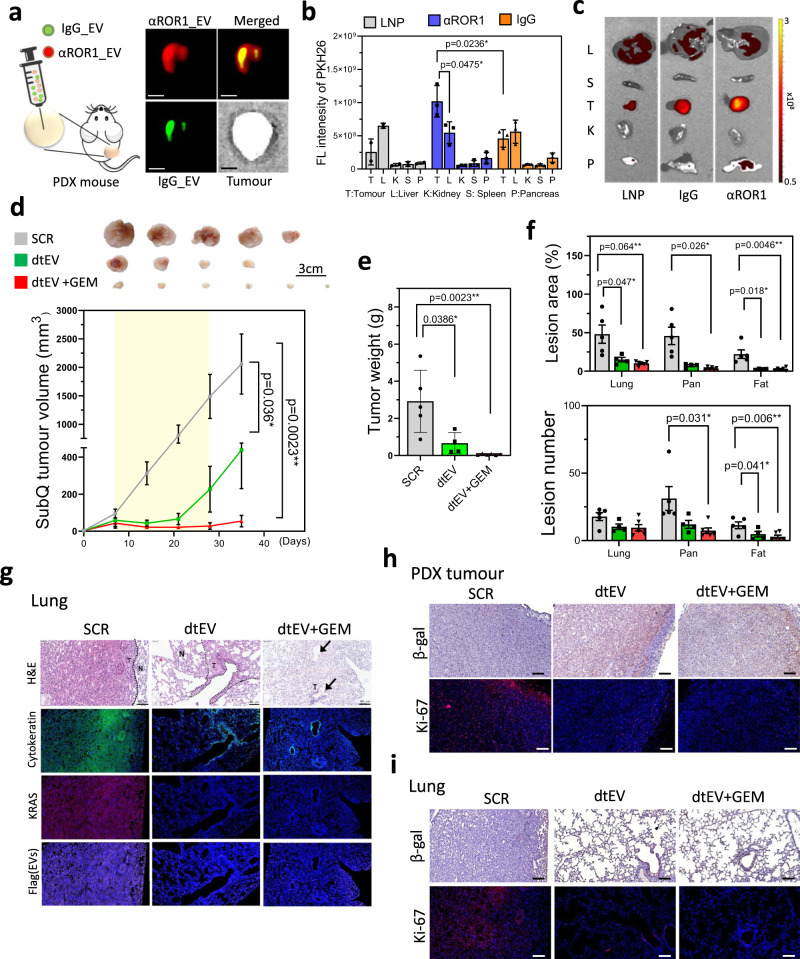
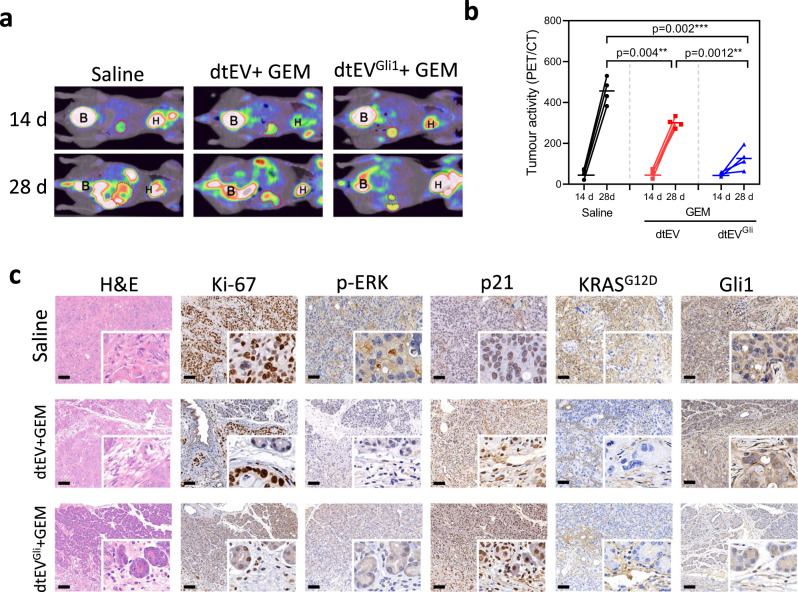
Pancreatic ductal adenocarcinoma (PDAC) tumours carry multiple gene mutations and respond poorly to treatments. There is currently an unmet need for drug carriers that can deliver multiple gene cargoes to target high solid tumour burden like PDAC. Here, we report a dual targeted extracellular vesicle (dtEV) carrying high loads of therapeutic RNA that effectively suppresses large PDAC tumours in mice. The EV surface contains a CD64 protein that has a tissue targeting peptide and a humanized monoclonal antibody. Cells sequentially transfected with plasmid DNAs encoding for the RNA and protein of interest by Transwell®-based asymmetric cell electroporation release abundant targeted EVs with high RNA loading. Together with a low dose chemotherapy drug, Gemcitabine, dtEVs suppress large orthotopic PANC-1 and patient derived xenograft tumours and metastasis in mice and extended animal survival. Our work presents a clinically accessible and scalable way to produce abundant EVs for delivering multiple gene cargoes to large solid tumours.
© 2023. Springer Nature Limited.
Conflict of interest statement
L.J.L. and A.S.L. are shareholders at Spot Biosystems Ltd. All other co-authors declare no competing interests.
Figures
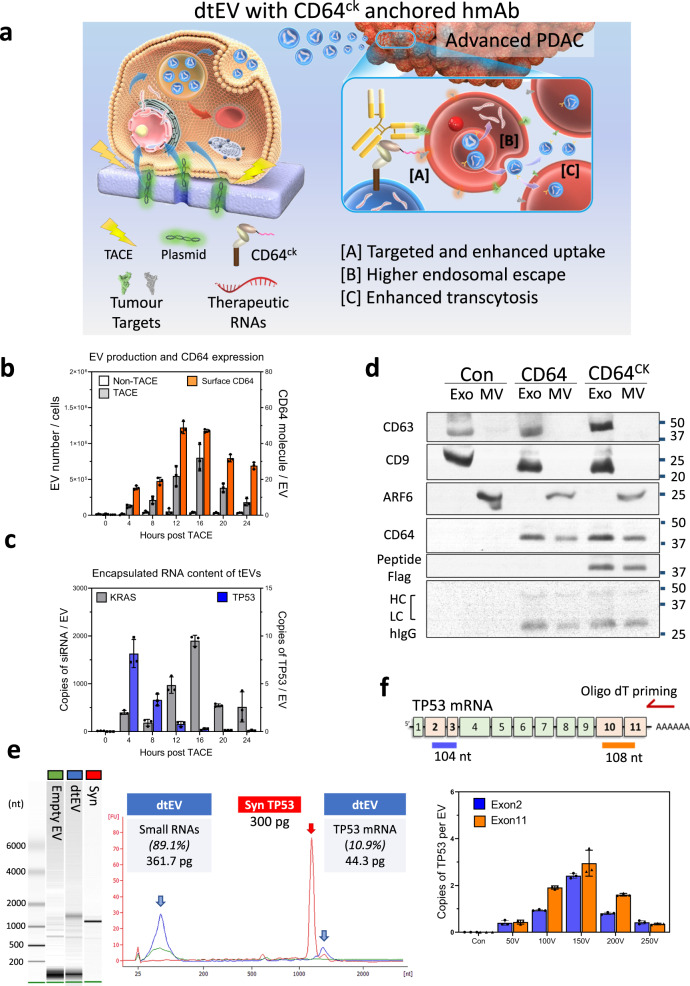
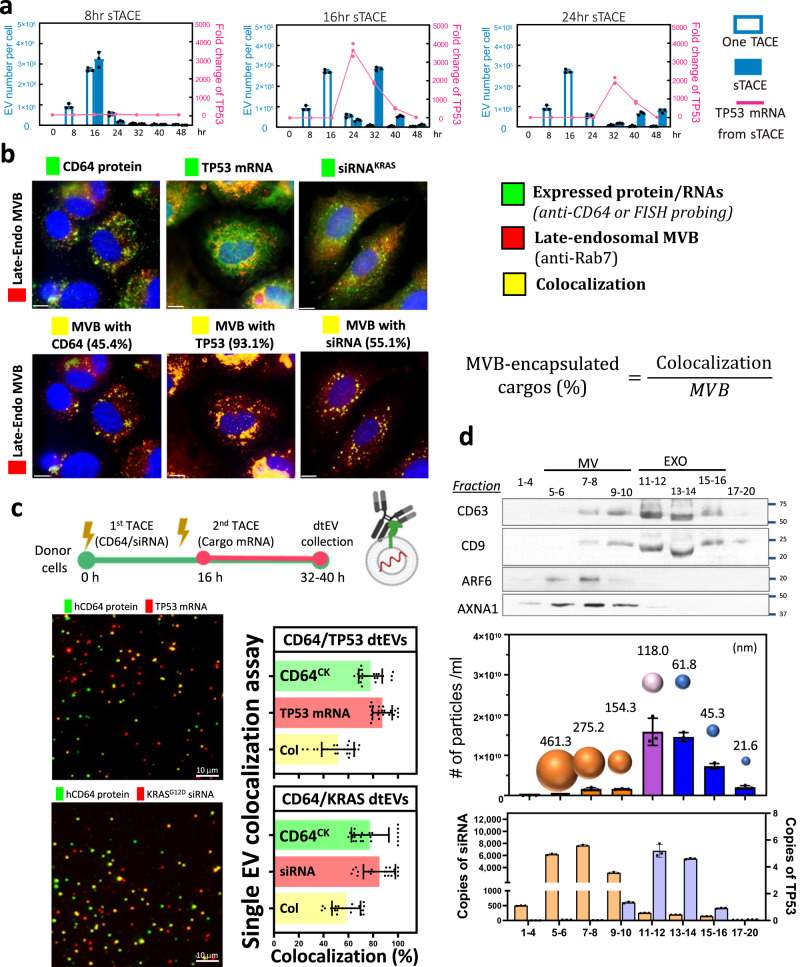
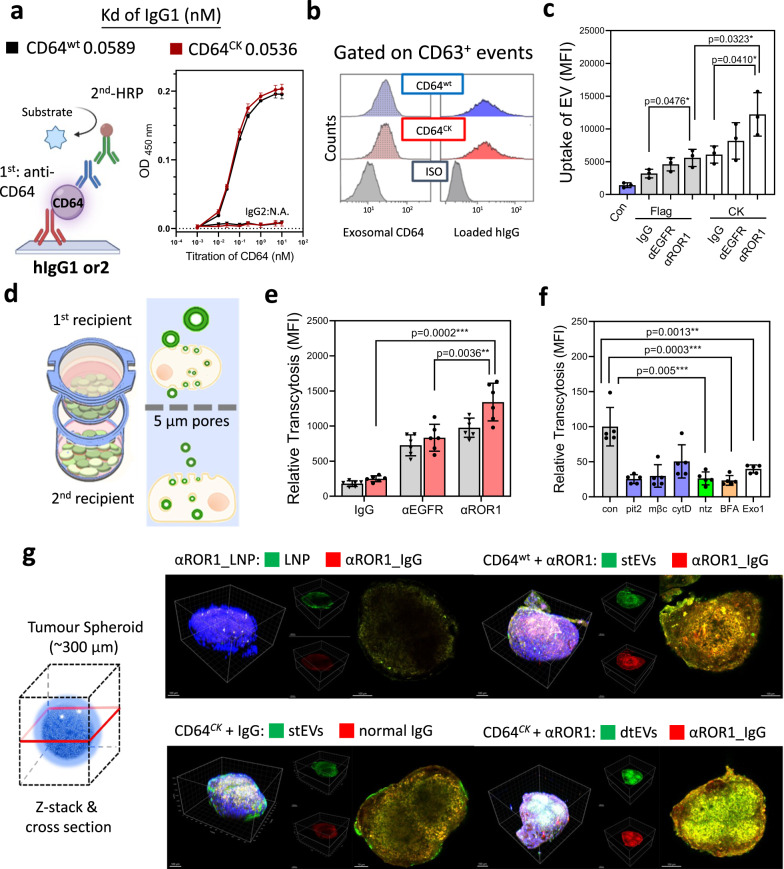
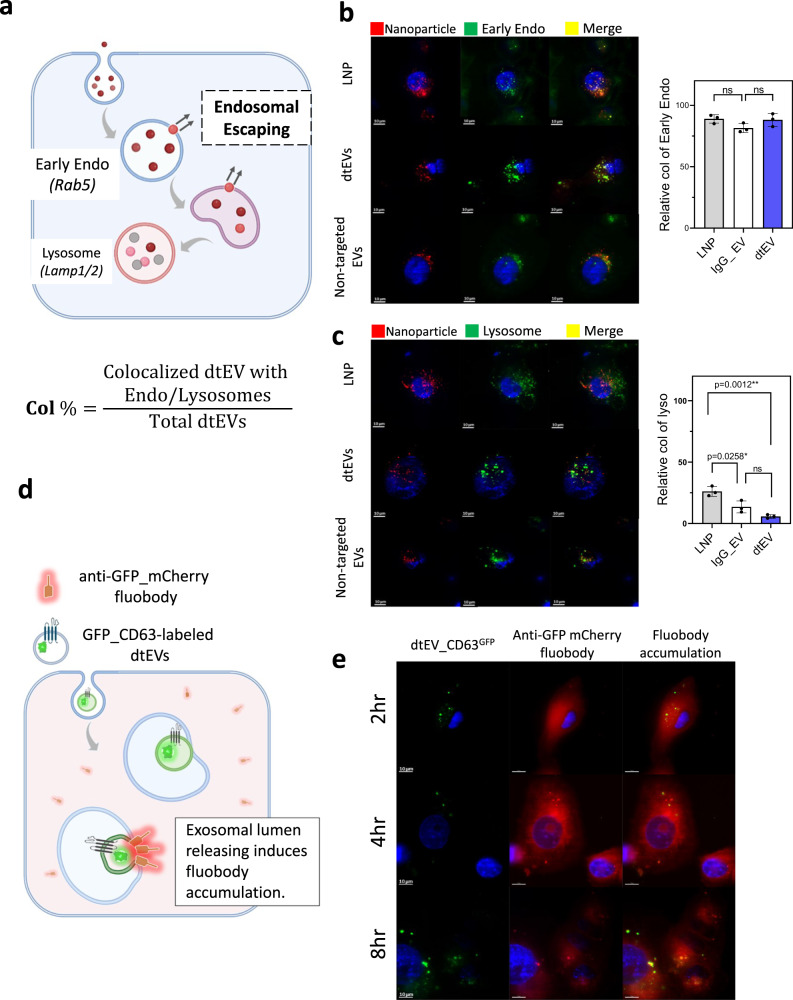
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
