

Pursuing excitonic energy transfer with programmable DNA-based optical breadboards
- PMID: 37872857
- PMCID: PMC10642627
- DOI: 10.1039/d0cs00936a
Pursuing excitonic energy transfer with programmable DNA-based optical breadboards
Abstract
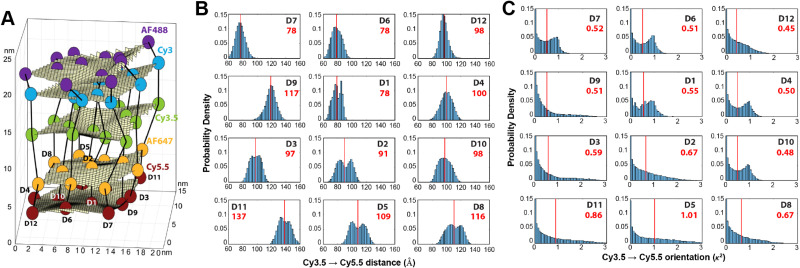
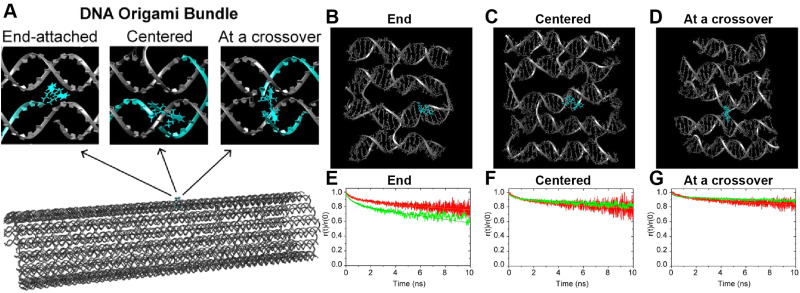
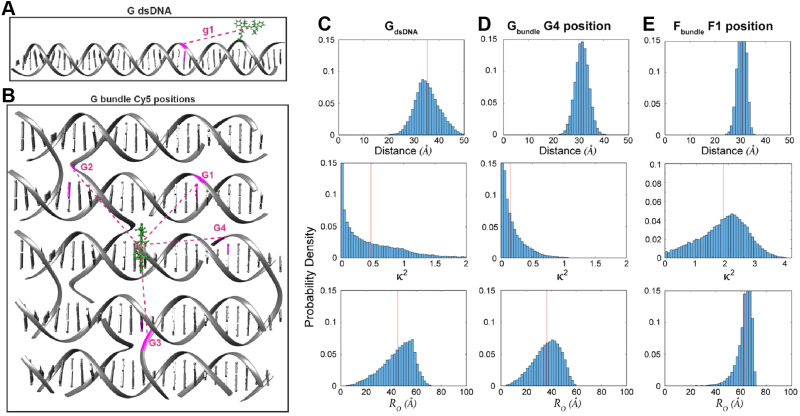
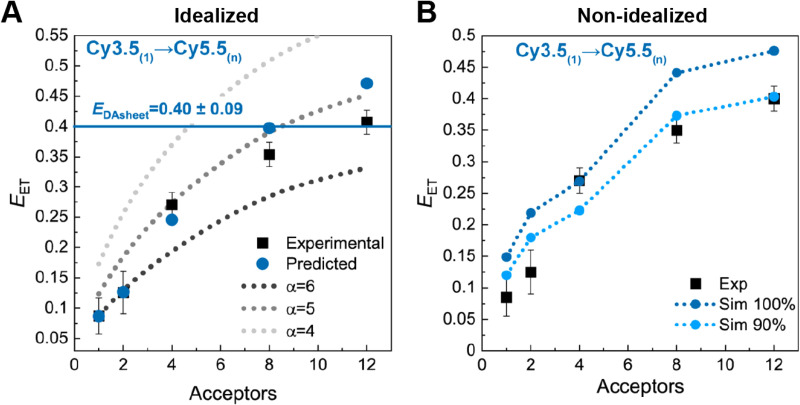
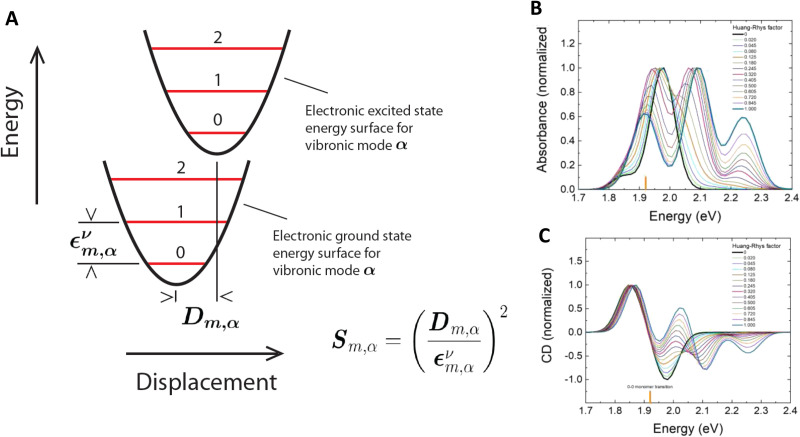
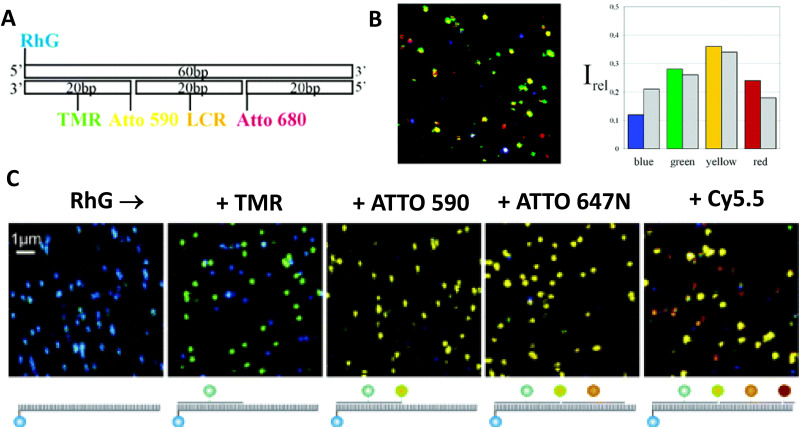
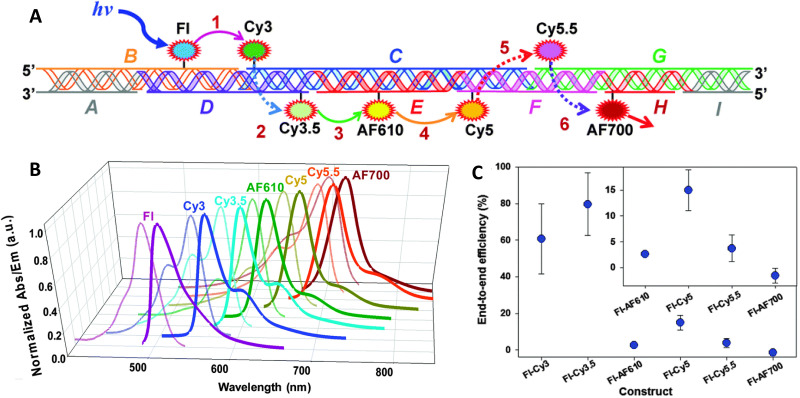
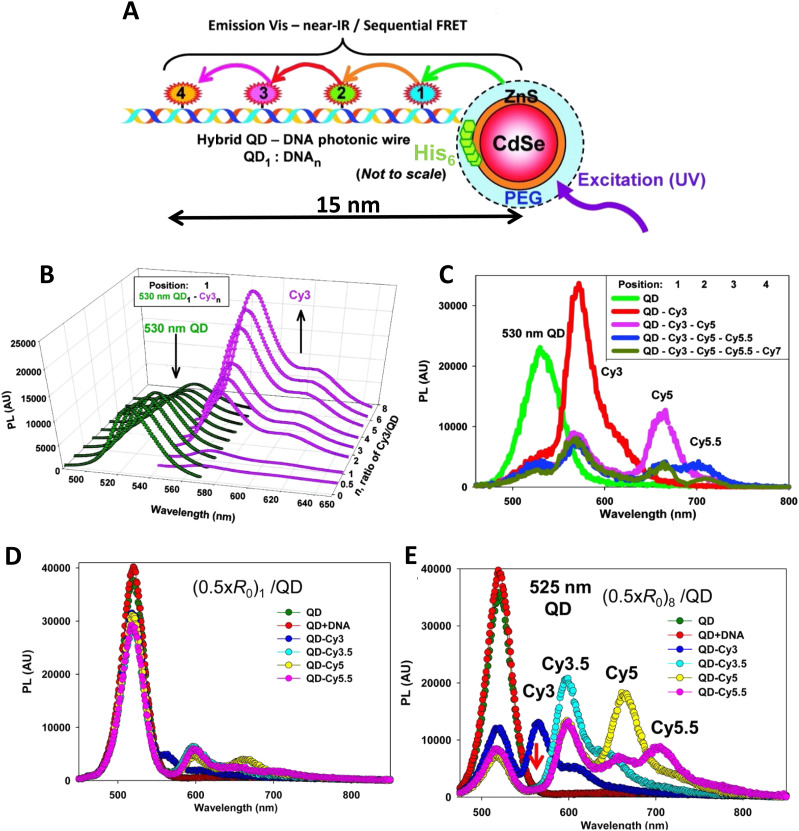
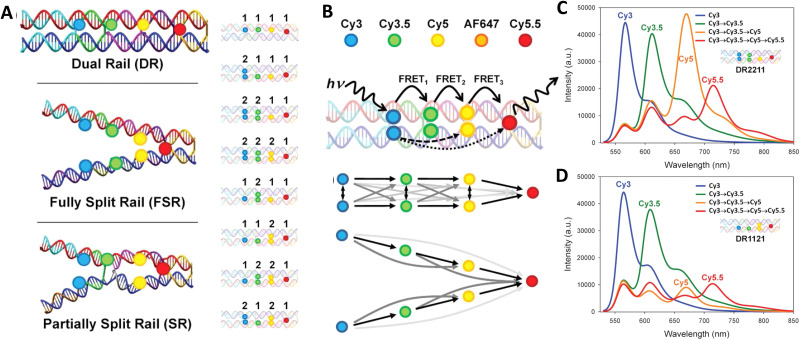
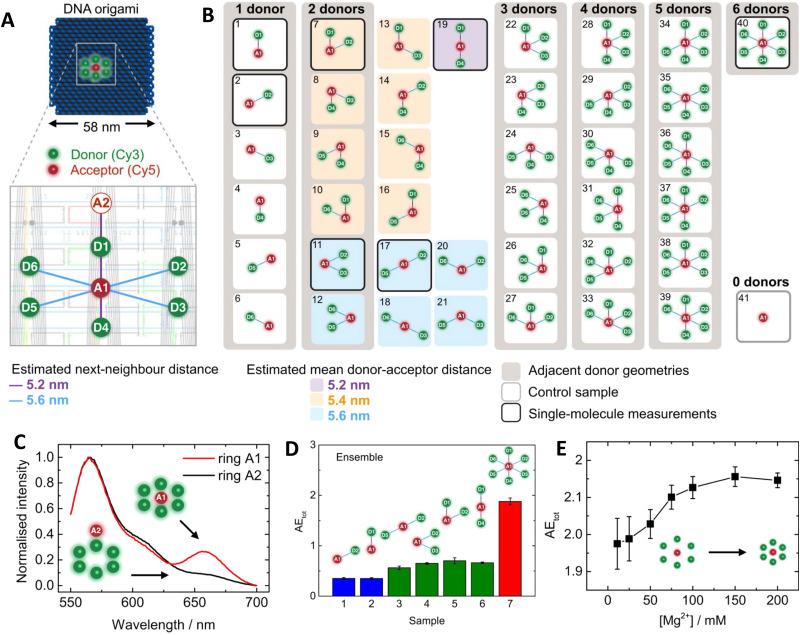
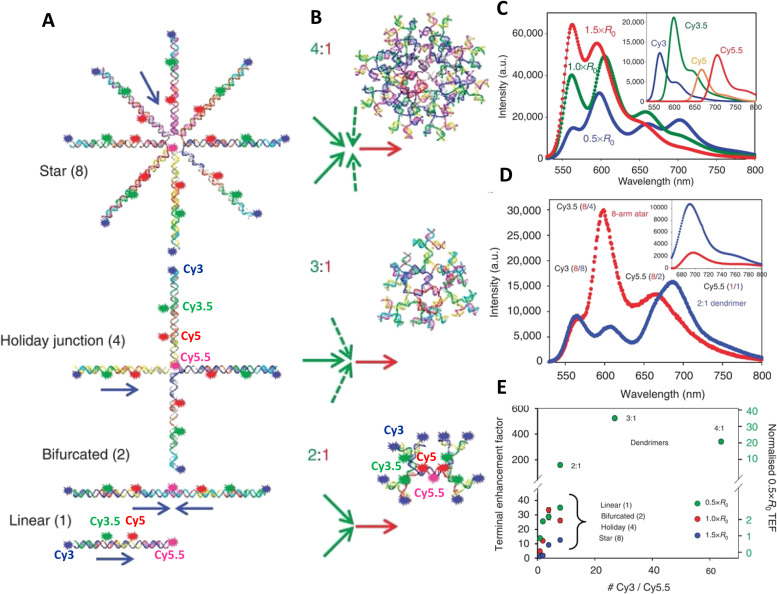
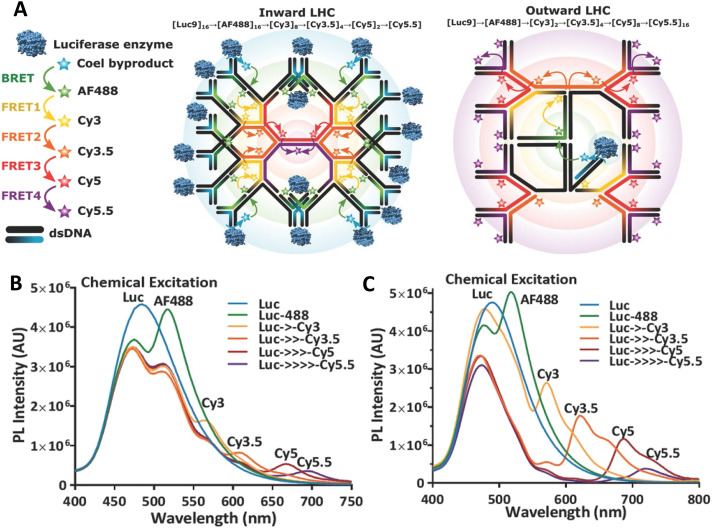
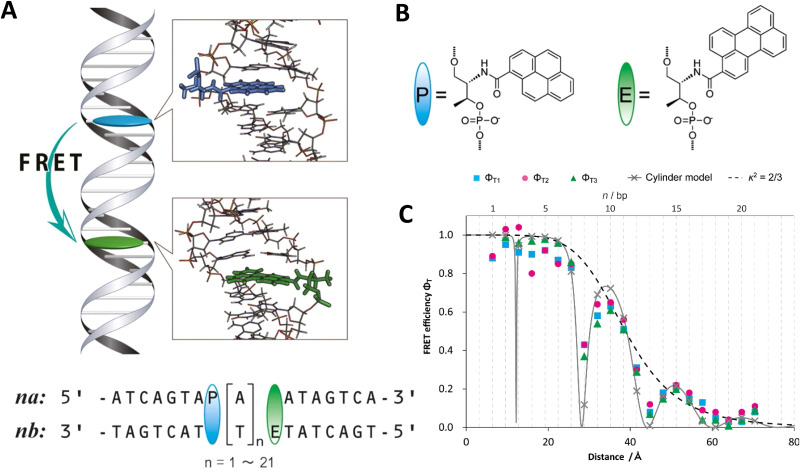
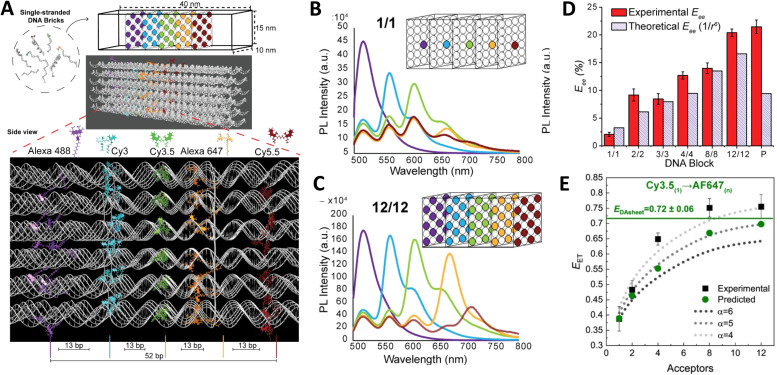
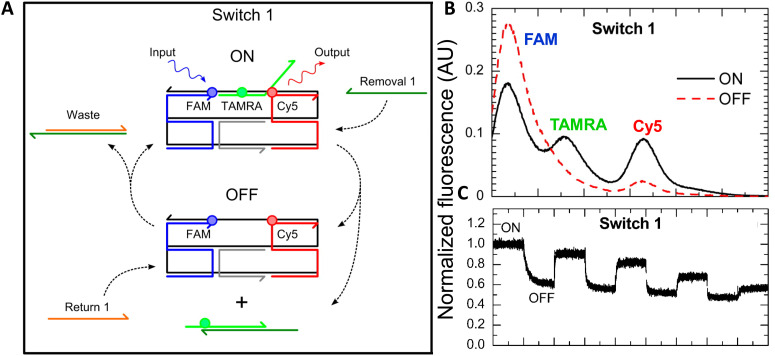
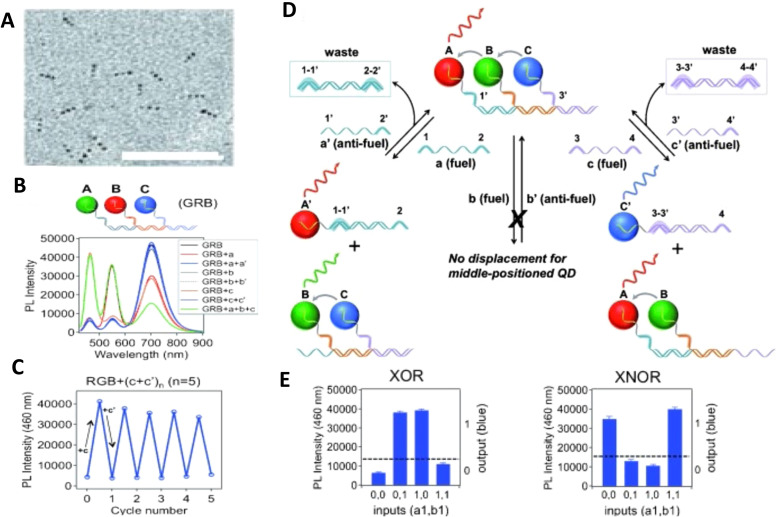
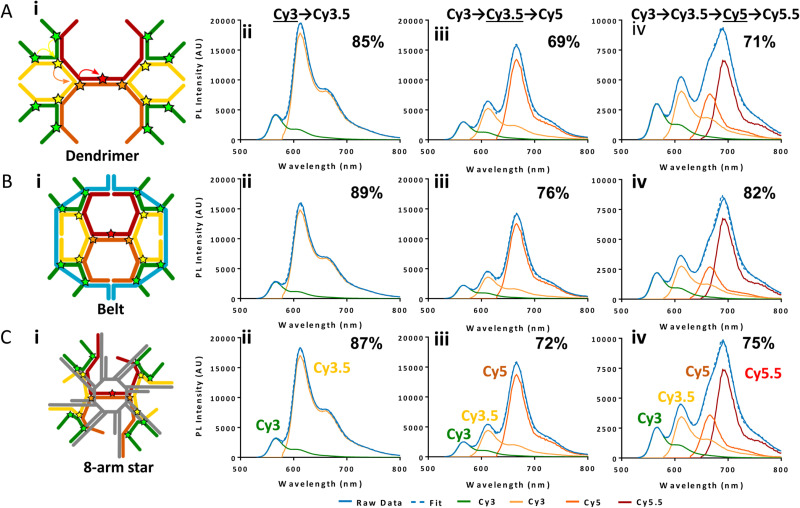
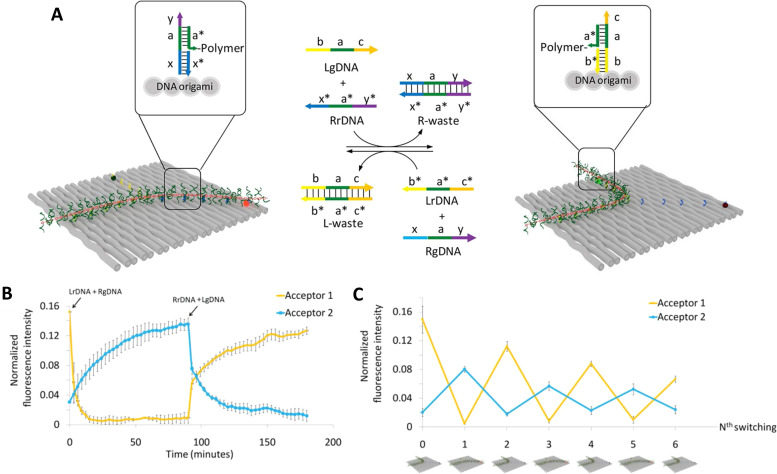
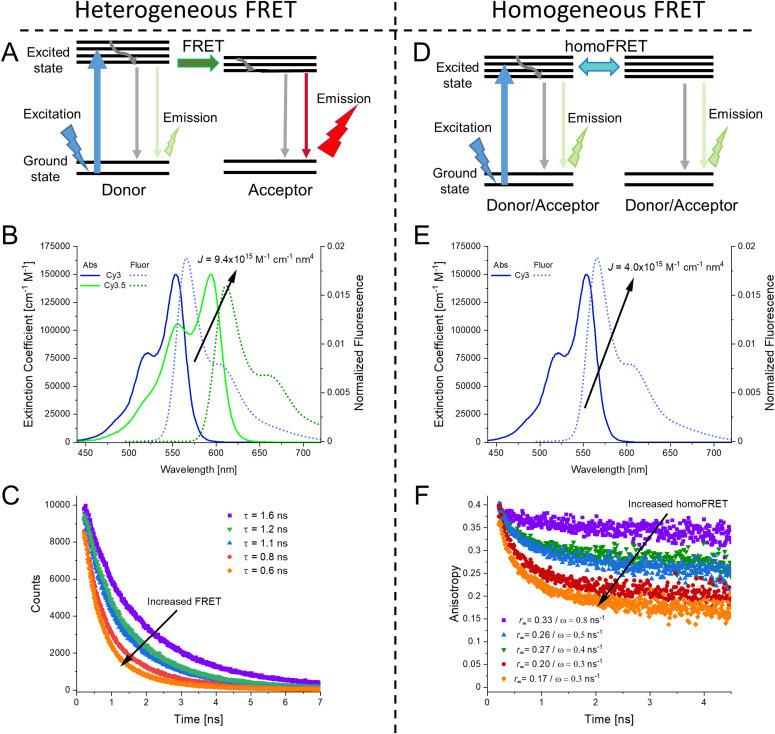
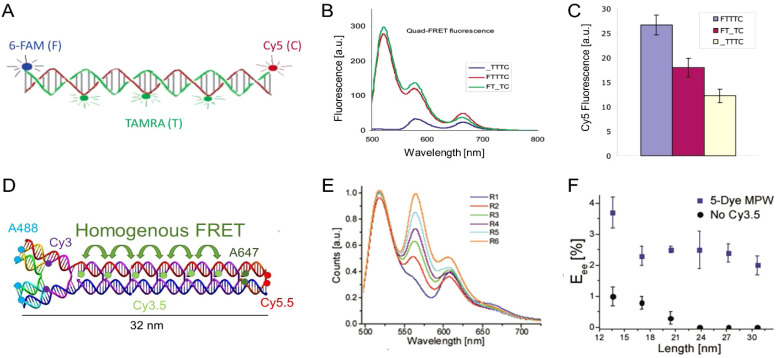
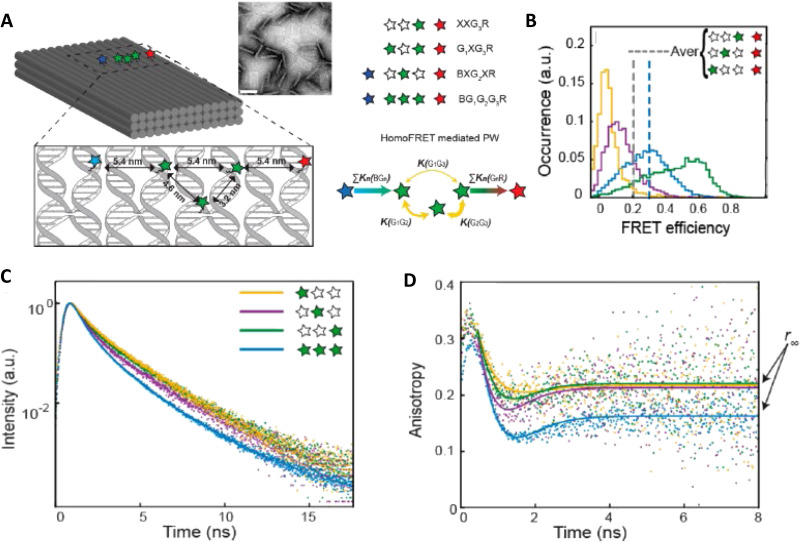
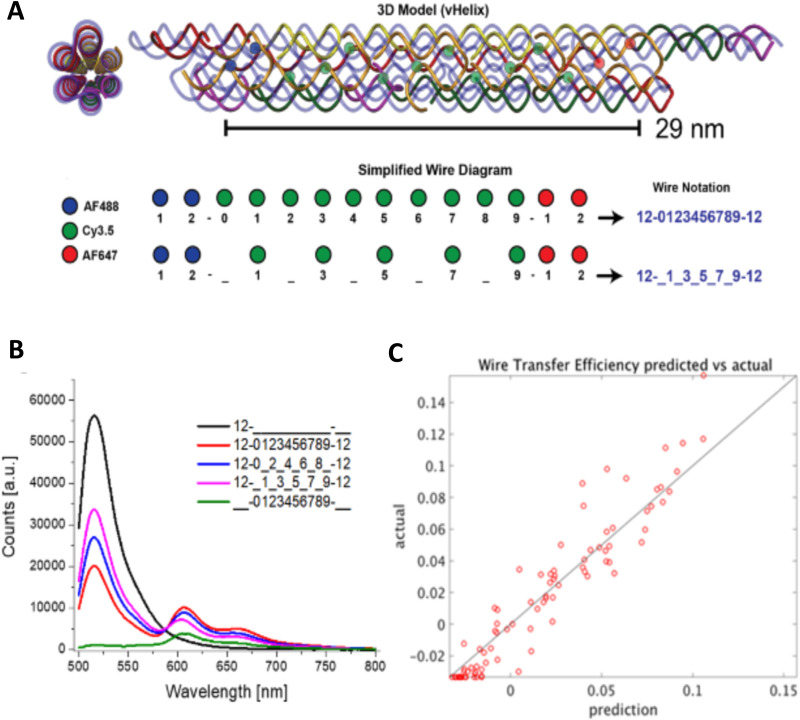
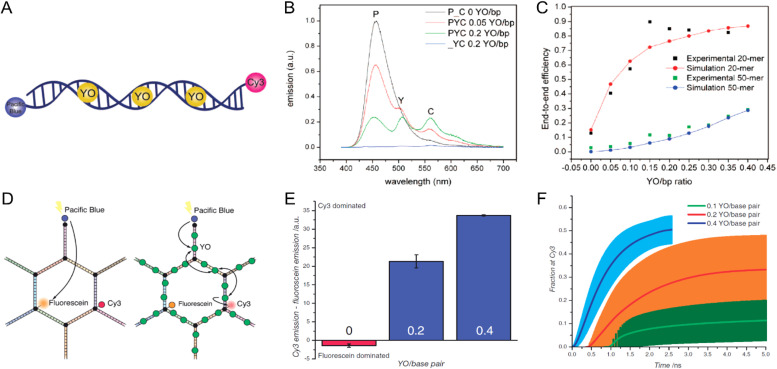
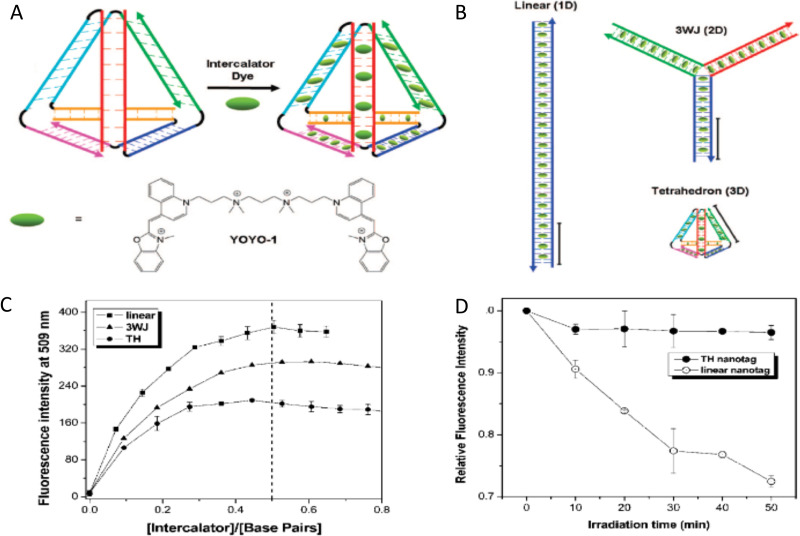
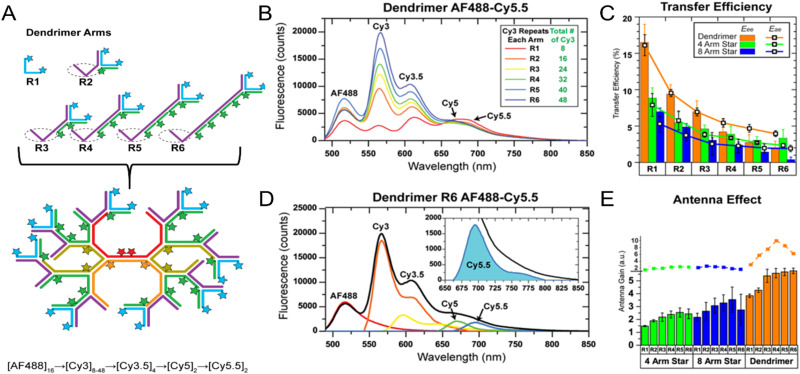
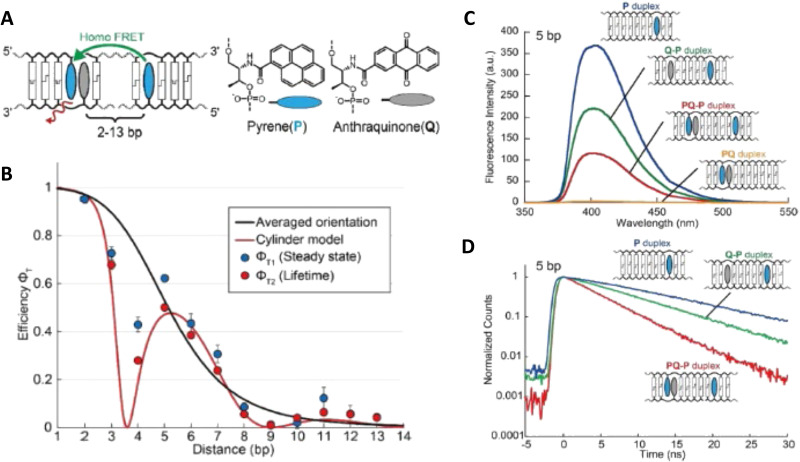
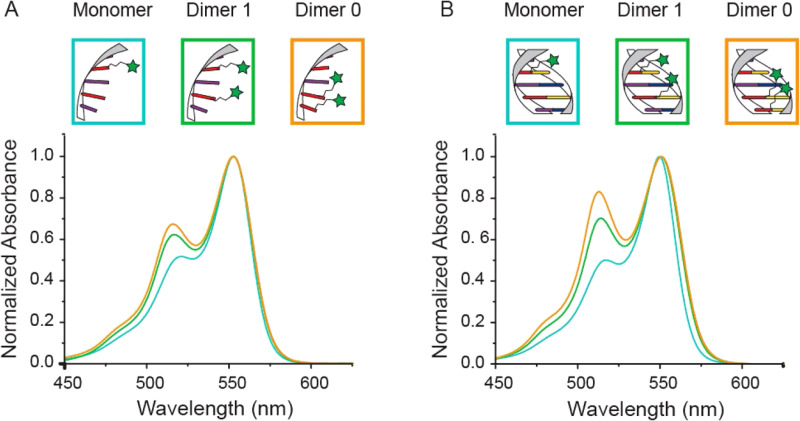
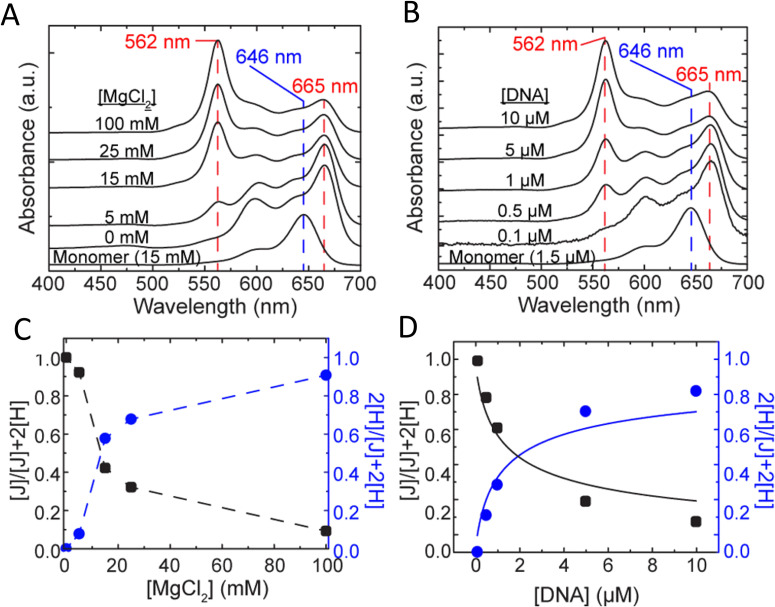
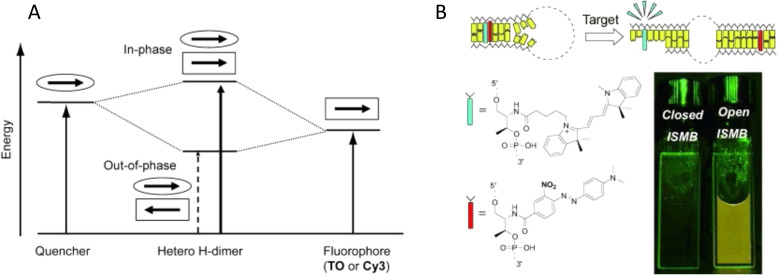
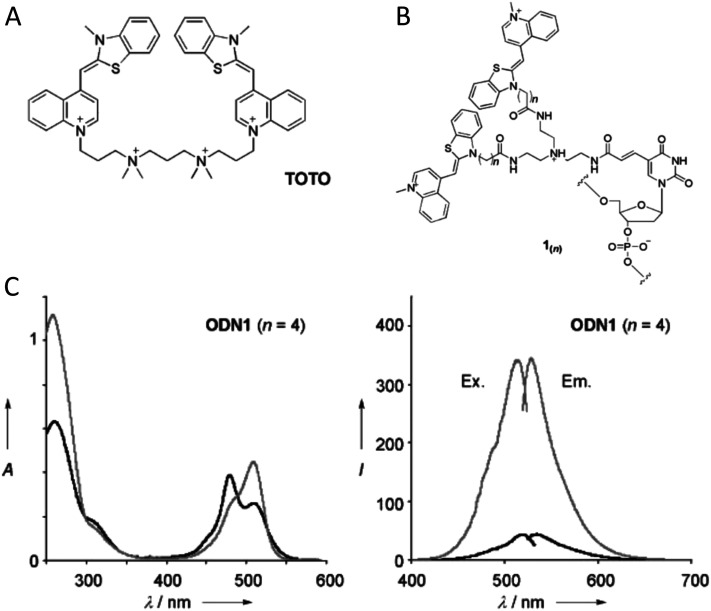
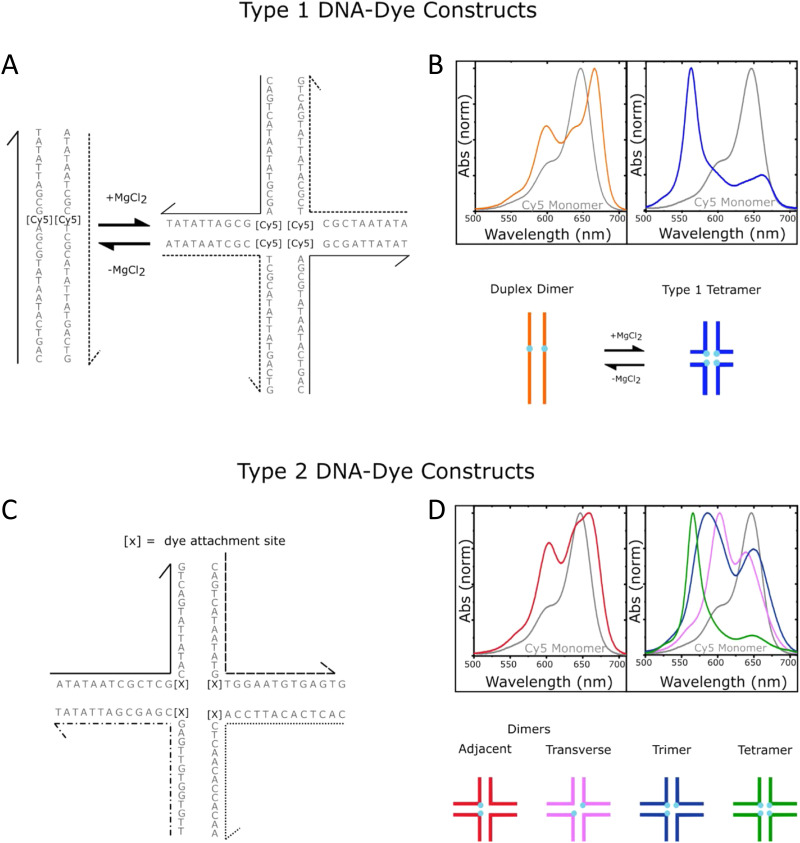
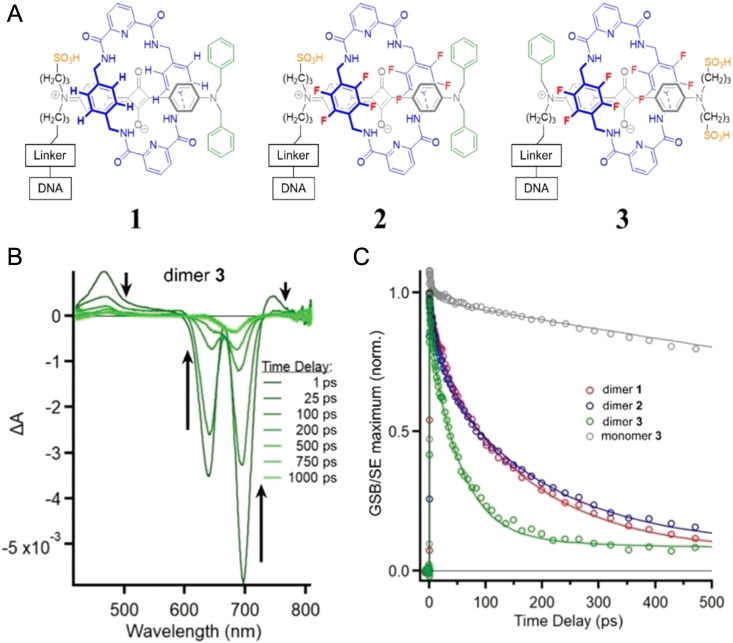
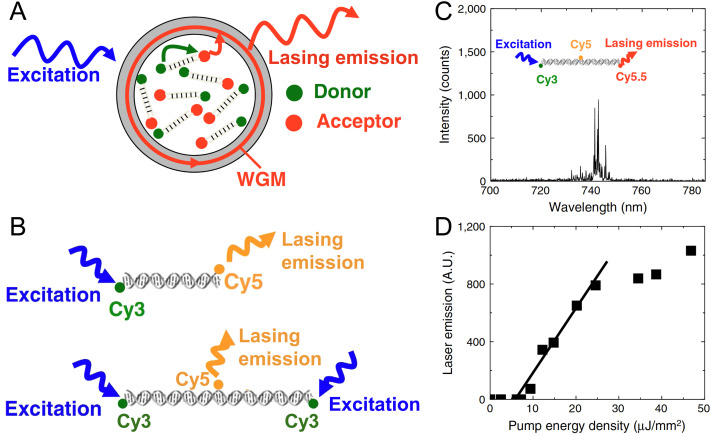
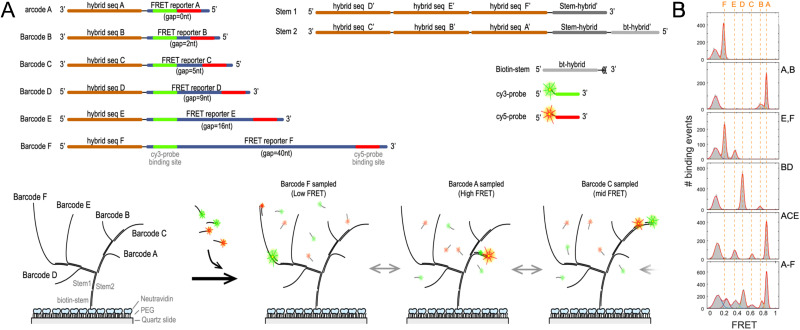
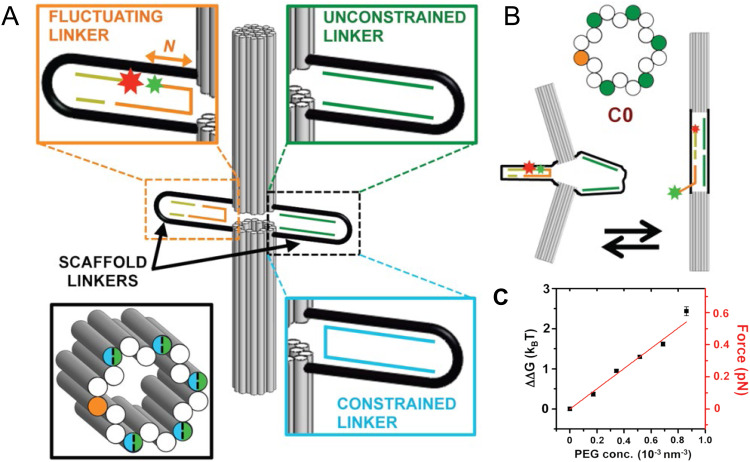
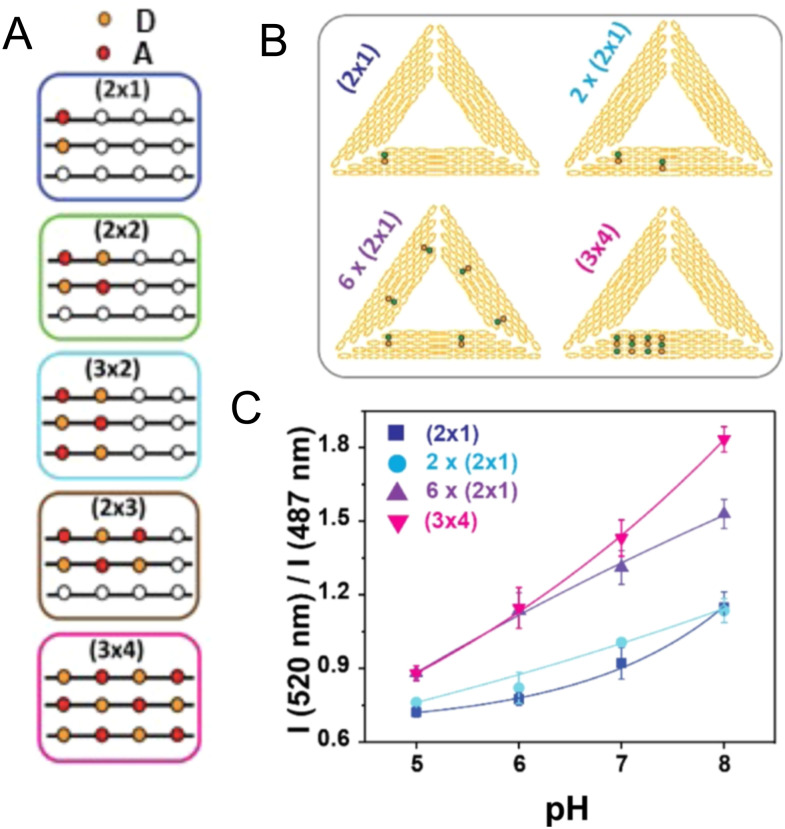
DNA nanotechnology has now enabled the self-assembly of almost any prescribed 3-dimensional nanoscale structure in large numbers and with high fidelity. These structures are also amenable to site-specific modification with a variety of small molecules ranging from drugs to reporter dyes. Beyond obvious application in biotechnology, such DNA structures are being pursued as programmable nanoscale optical breadboards where multiple different/identical fluorophores can be positioned with sub-nanometer resolution in a manner designed to allow them to engage in multistep excitonic energy-transfer (ET) via Förster resonance energy transfer (FRET) or other related processes. Not only is the ability to create such complex optical structures unique, more importantly, the ability to rapidly redesign and prototype almost all structural and optical analogues in a massively parallel format allows for deep insight into the underlying photophysical processes. Dynamic DNA structures further provide the unparalleled capability to reconfigure a DNA scaffold on the fly in situ and thus switch between ET pathways within a given assembly, actively change its properties, and even repeatedly toggle between two states such as on/off. Here, we review progress in developing these composite materials for potential applications that include artificial light harvesting, smart sensors, nanoactuators, optical barcoding, bioprobes, cryptography, computing, charge conversion, and theranostics to even new forms of optical data storage. Along with an introduction into the DNA scaffolding itself, the diverse fluorophores utilized in these structures, their incorporation chemistry, and the photophysical processes they are designed to exploit, we highlight the evolution of DNA architectures implemented in the pursuit of increased transfer efficiency and the key lessons about ET learned from each iteration. We also focus on recent and growing efforts to exploit DNA as a scaffold for assembling molecular dye aggregates that host delocalized excitons as a test bed for creating excitonic circuits and accessing other quantum-like optical phenomena. We conclude with an outlook on what is still required to transition these materials from a research pursuit to application specific prototypes and beyond.
Conflict of interest statement
There are no conflicts to declare.
Figures

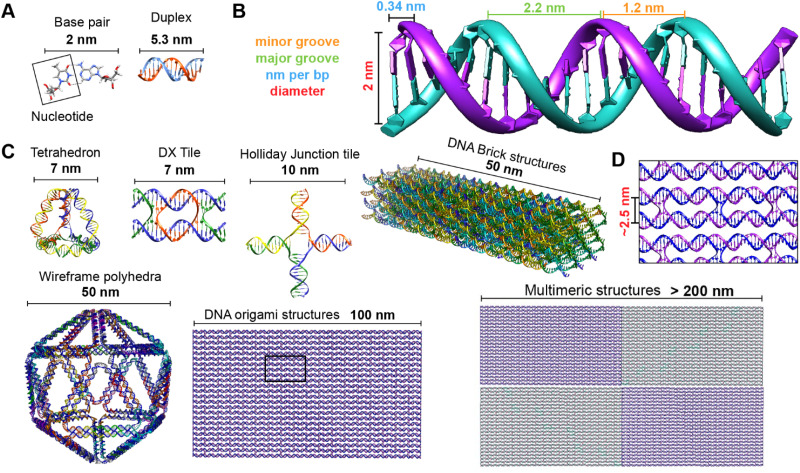
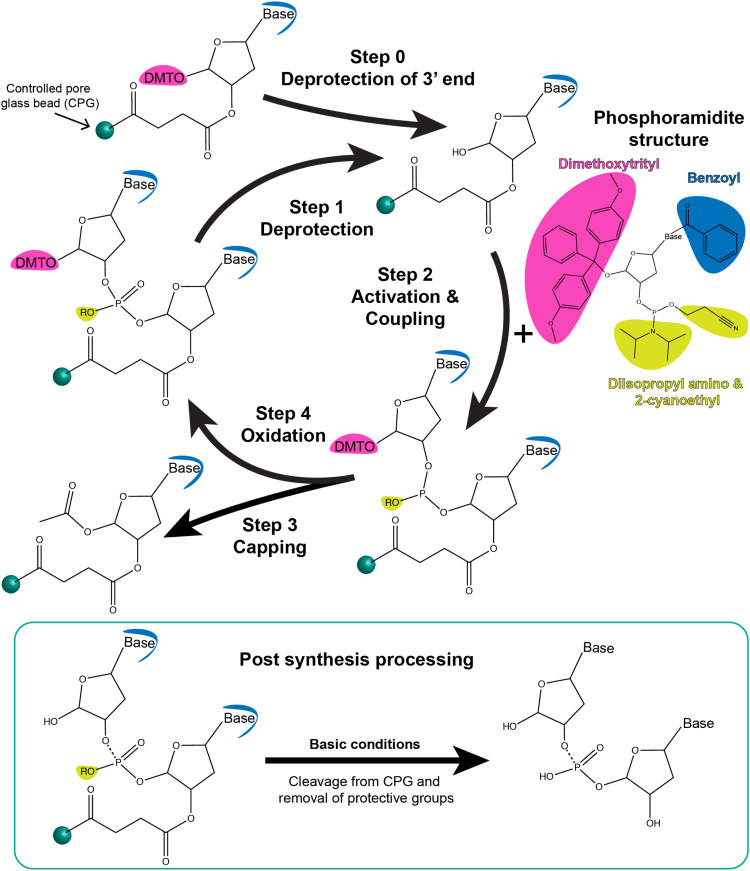
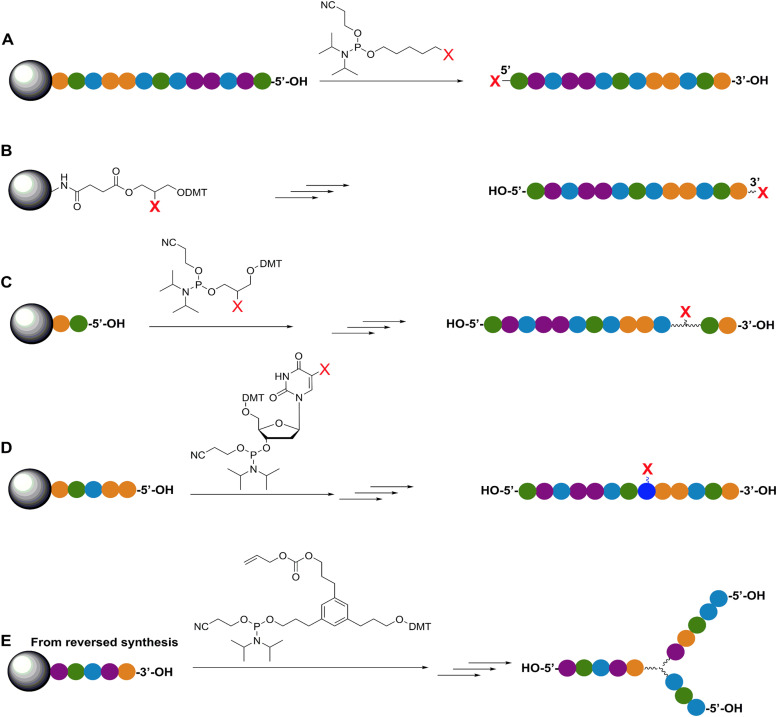
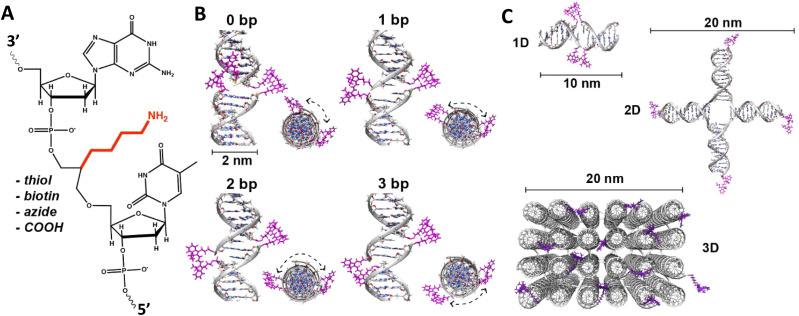
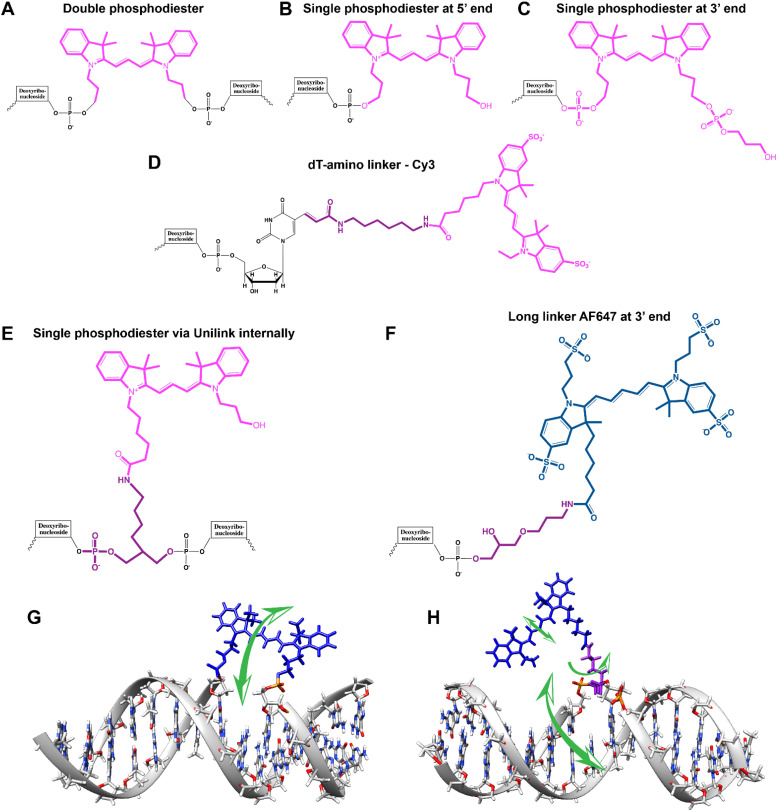
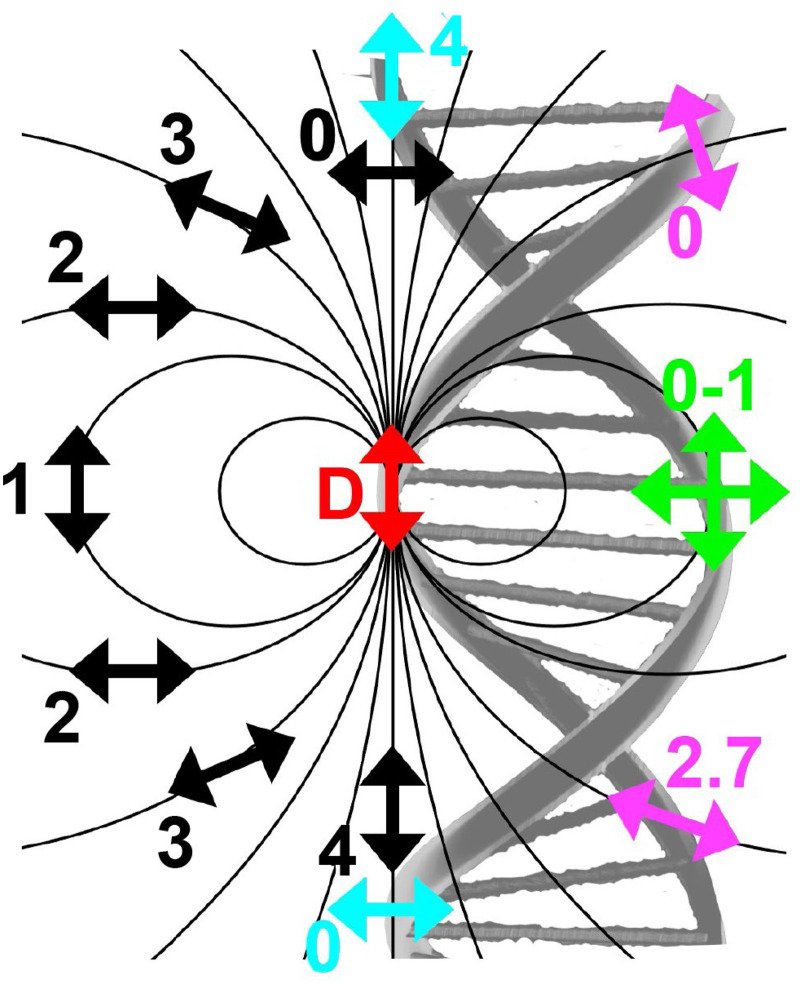
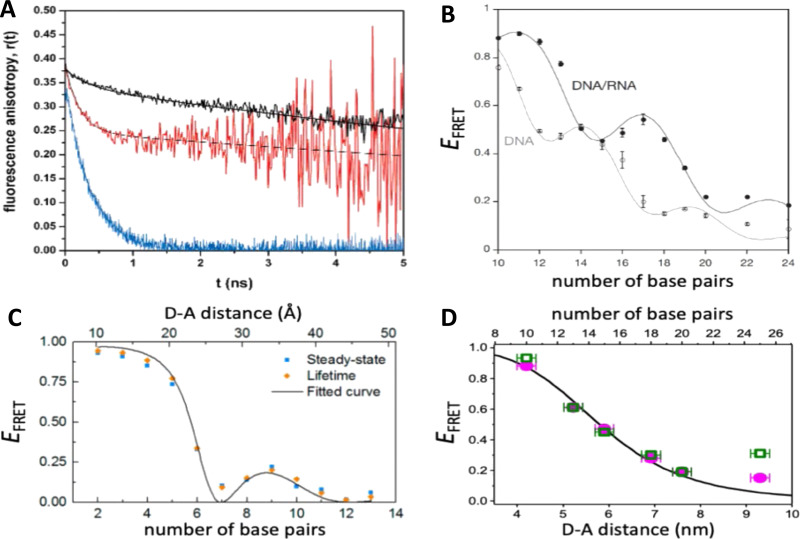
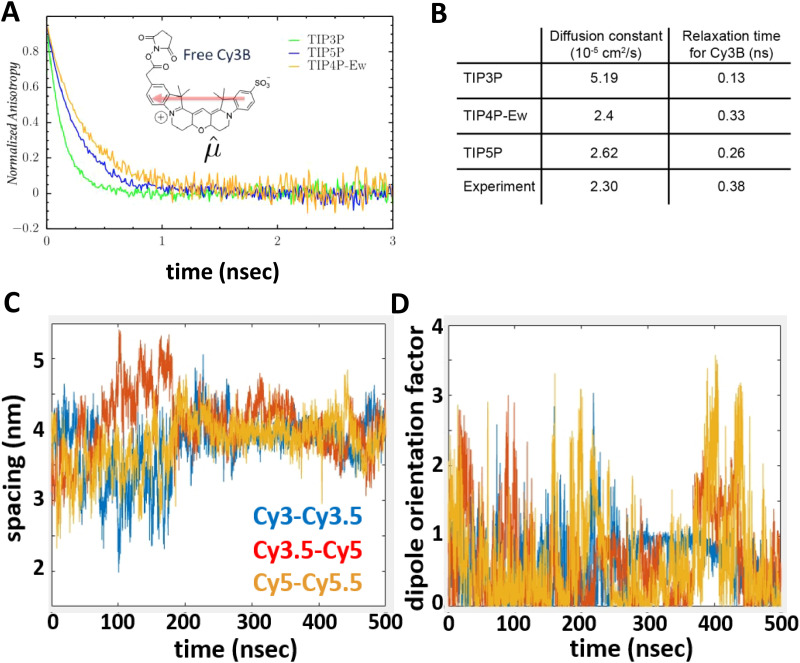
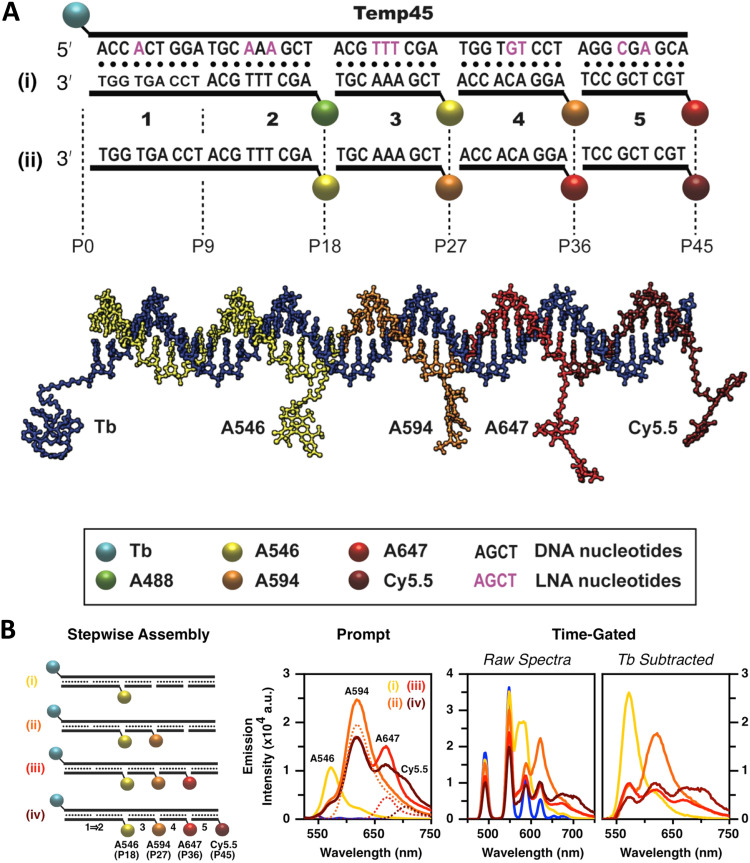
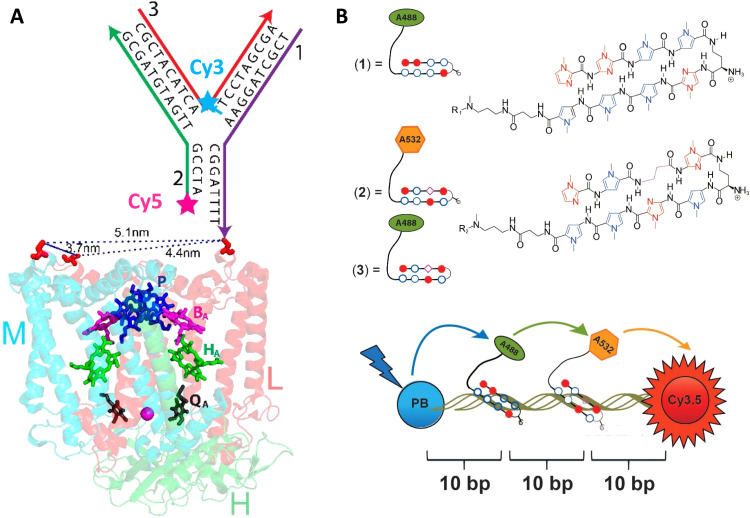
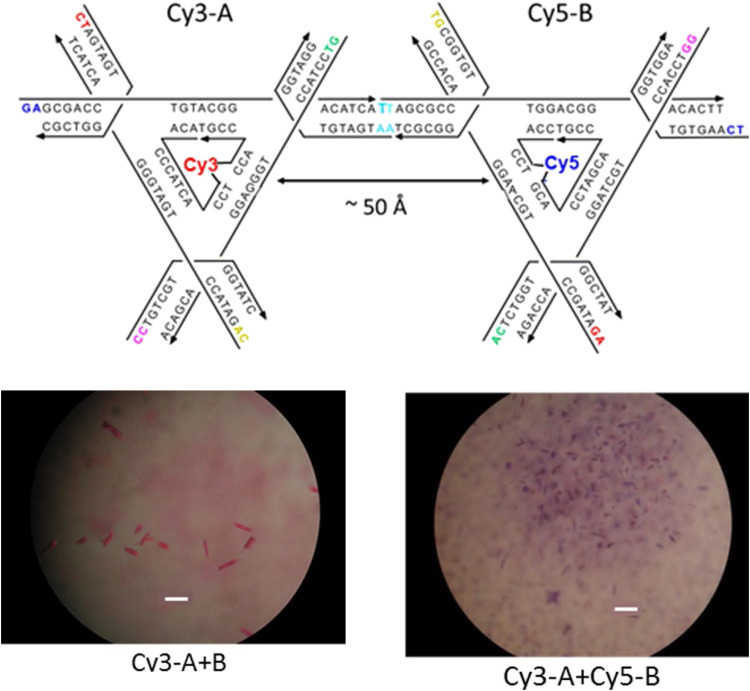
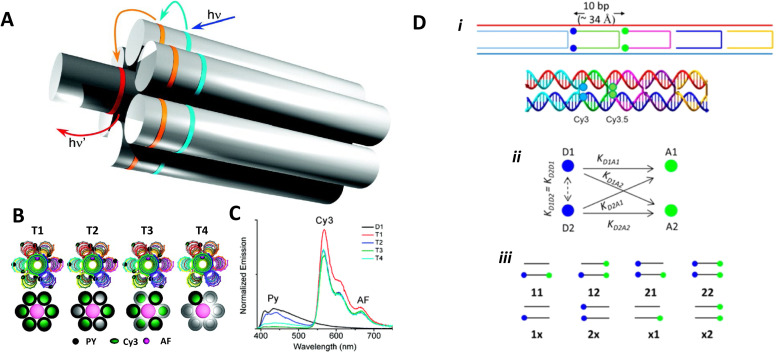
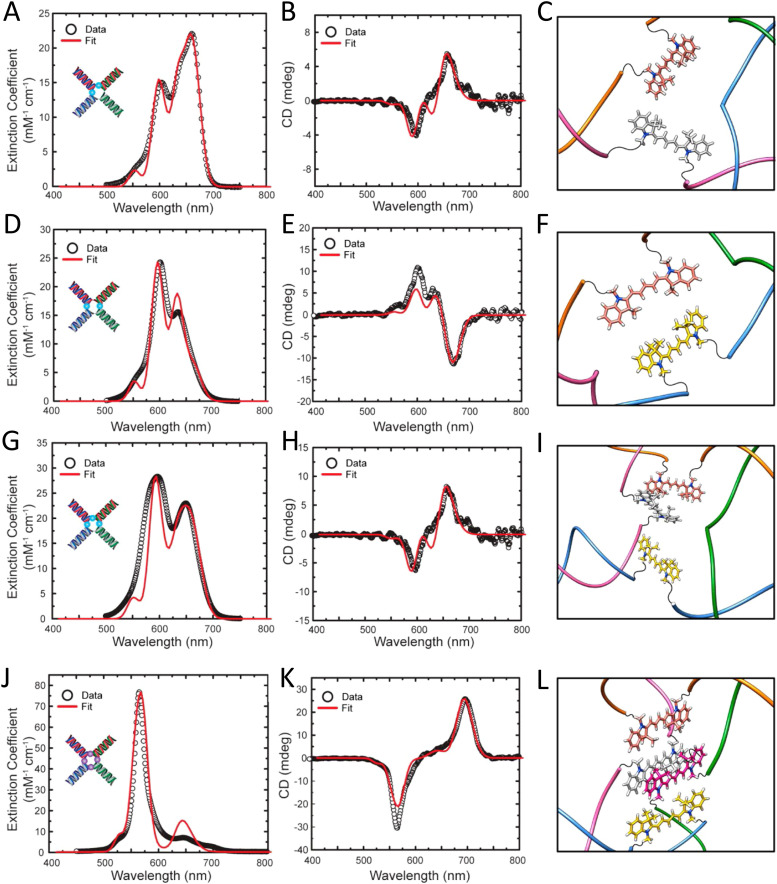
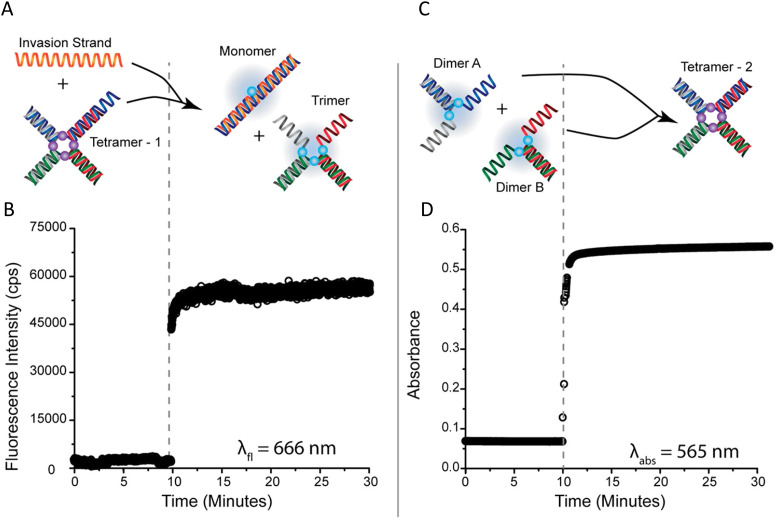
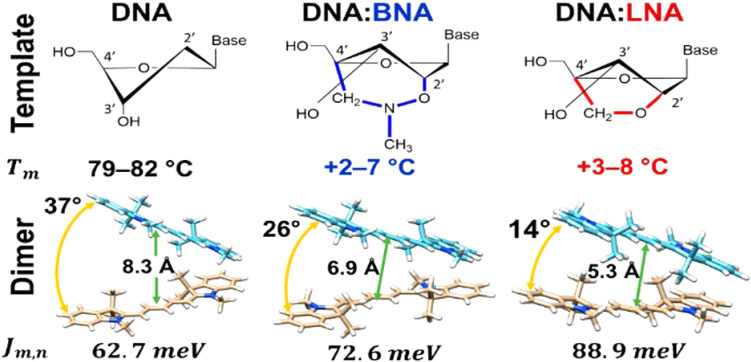
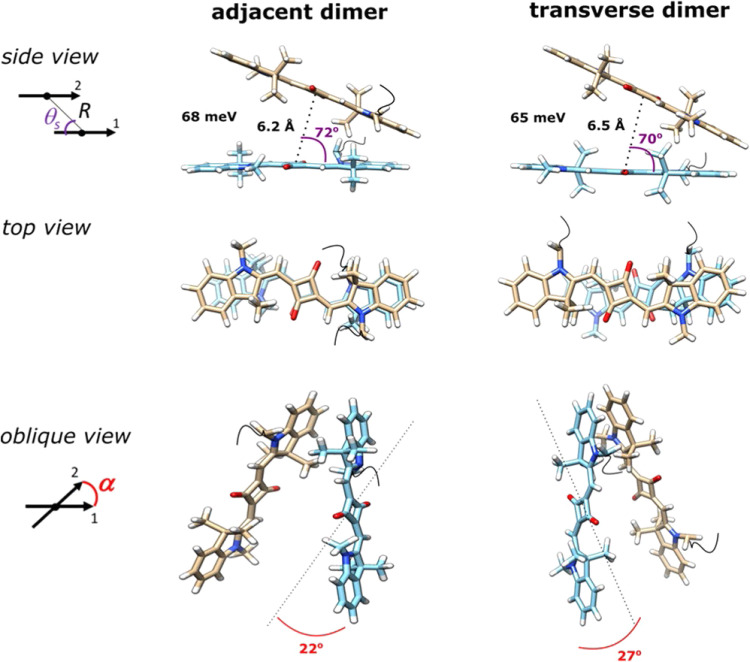
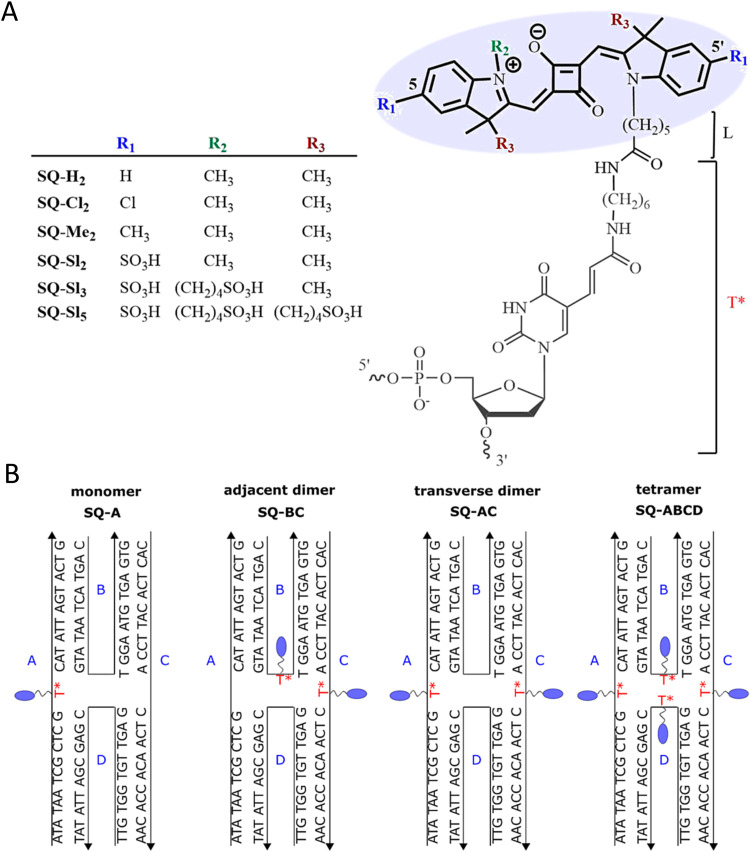
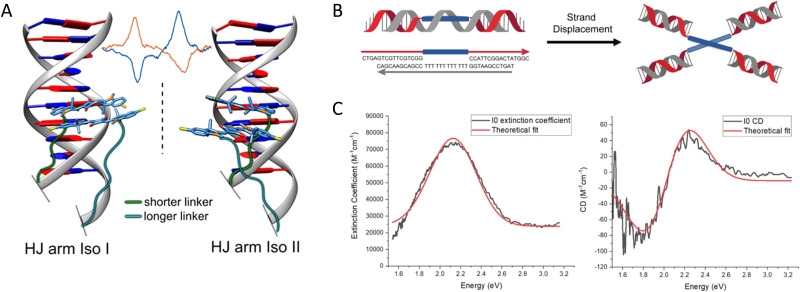
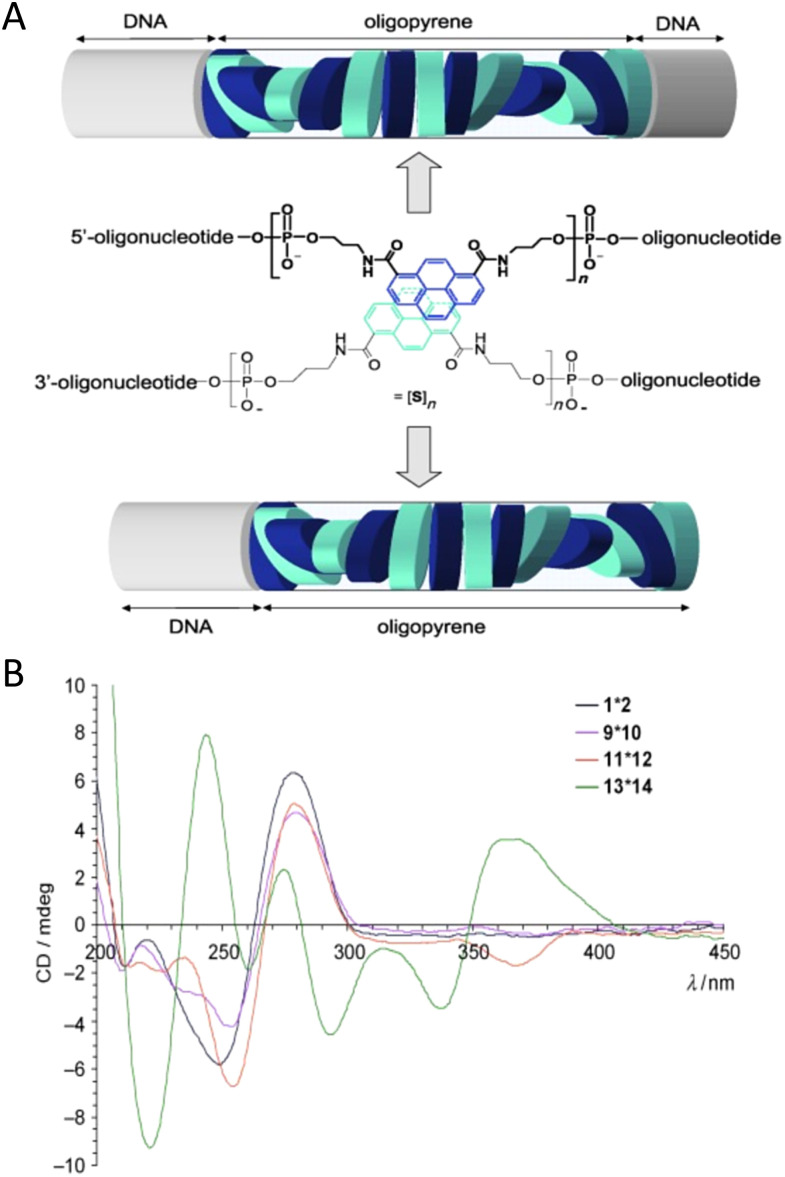
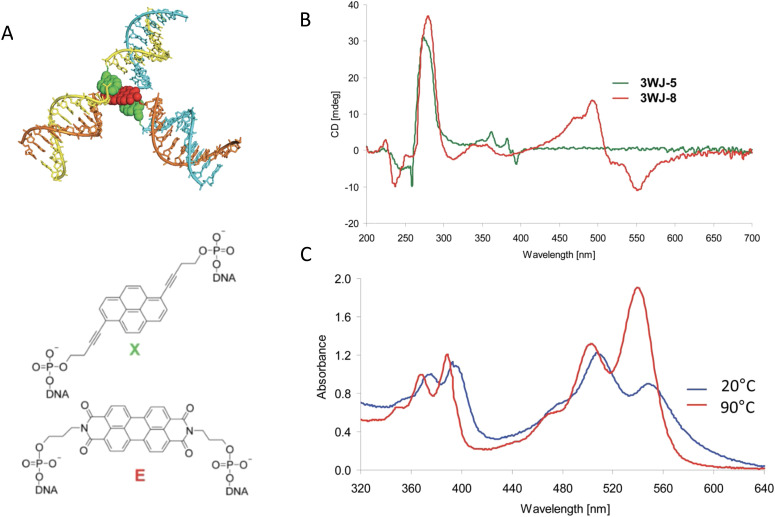
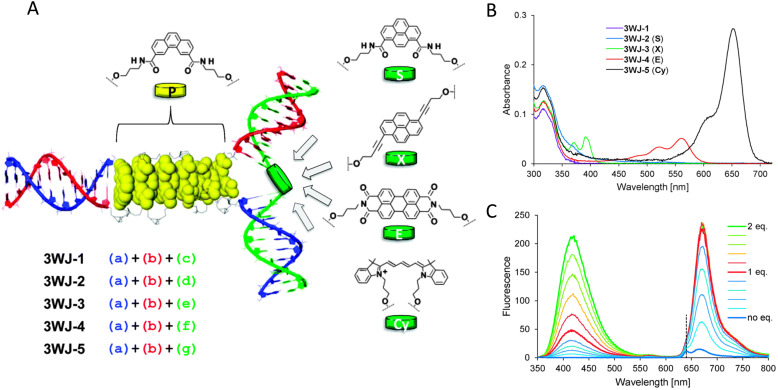
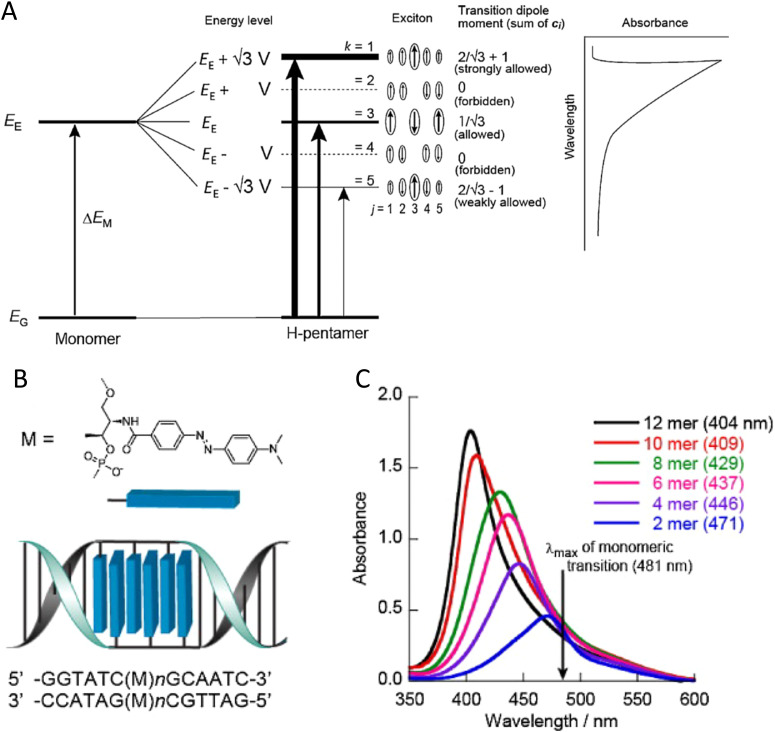
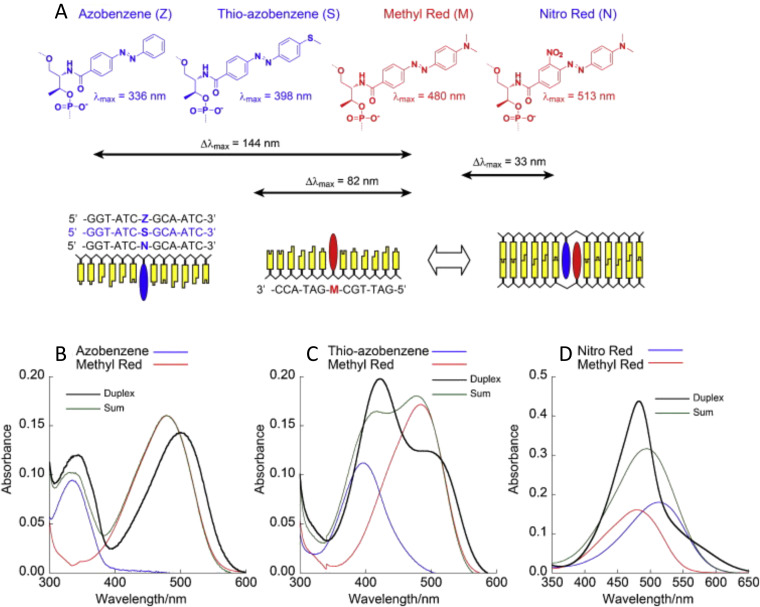
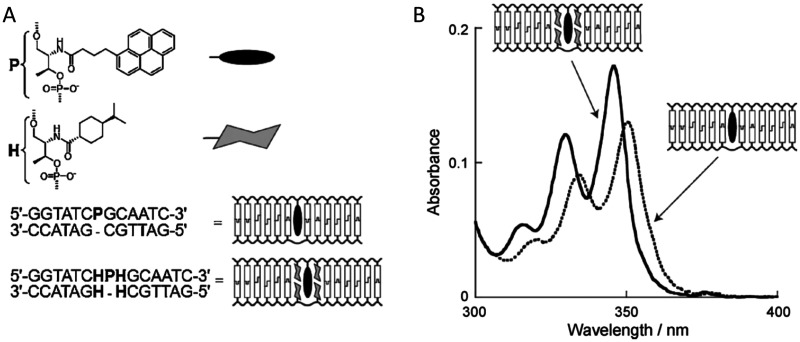
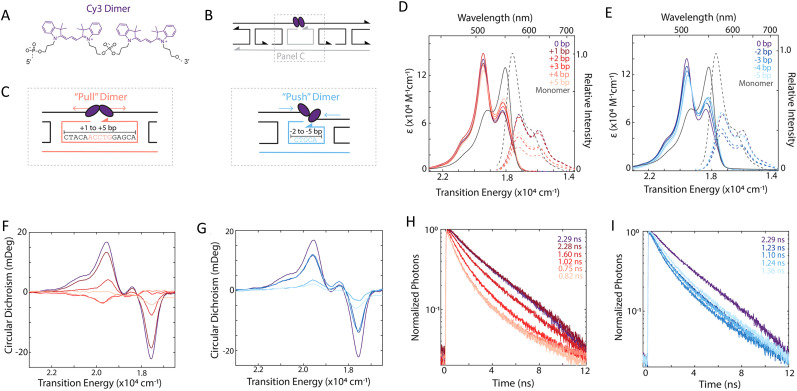
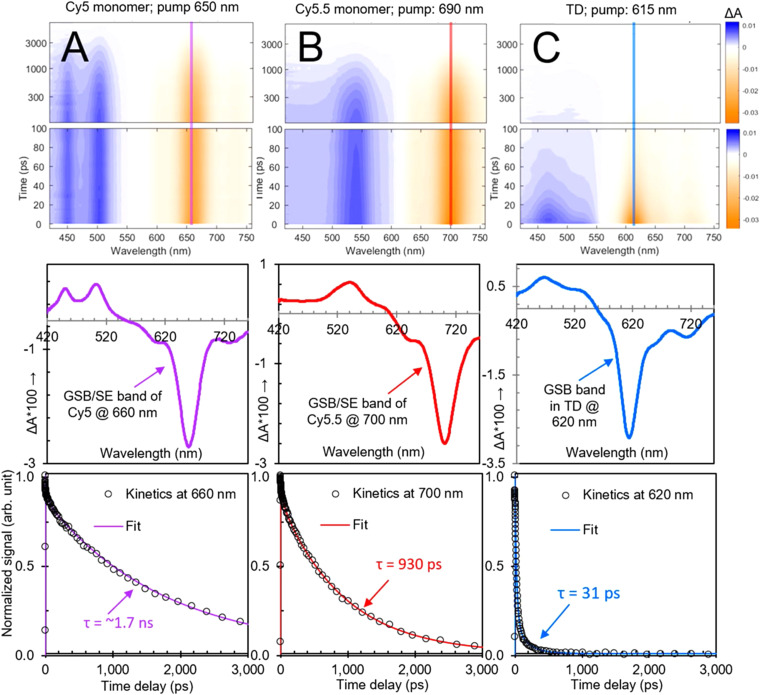
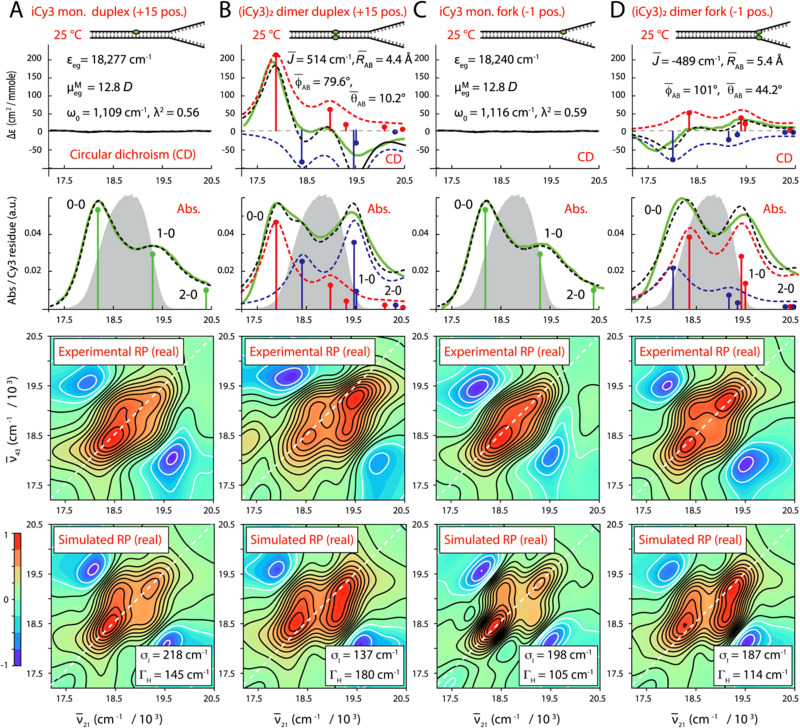
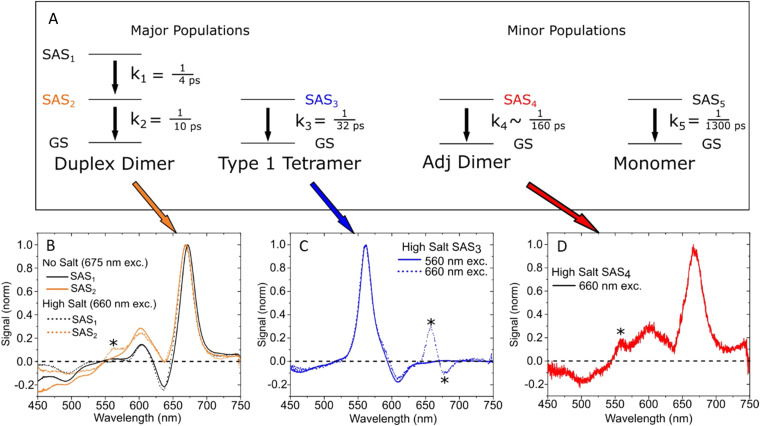
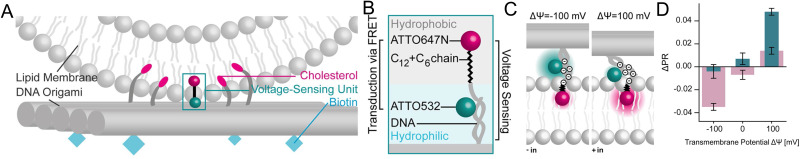
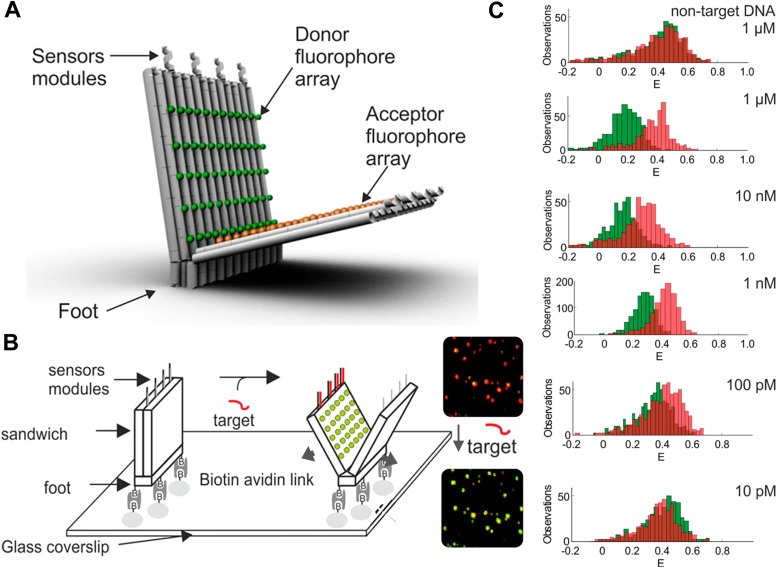
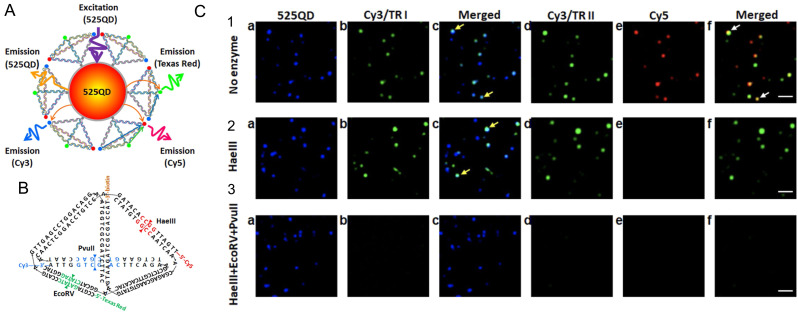
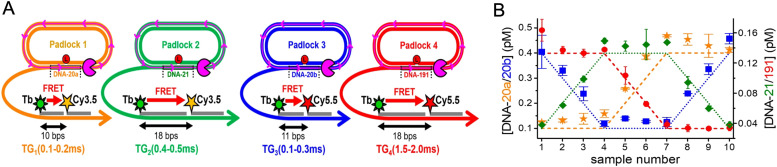
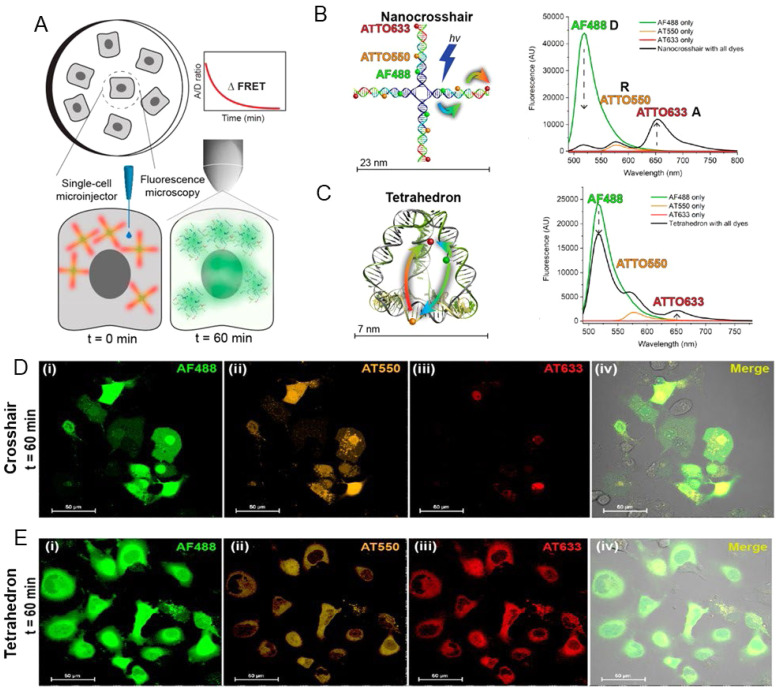
References
-
- Oh E. Fatemi F. K. Currie M. Delehanty J. B. Pons T. Fragola A. Leveque-Fort S. Goswami R. Susumu K. Huston A. L. Medintz I. L. Part. Part. Syst. Charact. 2013;30:453–466. doi: 10.1002/ppsc.201200140. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources