

This is a preprint.
Inferring B cell phylogenies from paired heavy and light chain BCR sequences with Dowser
- PMID: 37873135
- PMCID: PMC10592837
- DOI: 10.1101/2023.09.29.560187
Inferring B cell phylogenies from paired heavy and light chain BCR sequences with Dowser
Update in
-
Inferring B Cell Phylogenies from Paired H and L Chain BCR Sequences with Dowser.J Immunol. 2024 May 15;212(10):1579-1588. doi: 10.4049/jimmunol.2300851. J Immunol. 2024. PMID: 38557795 Free PMC article.
Abstract
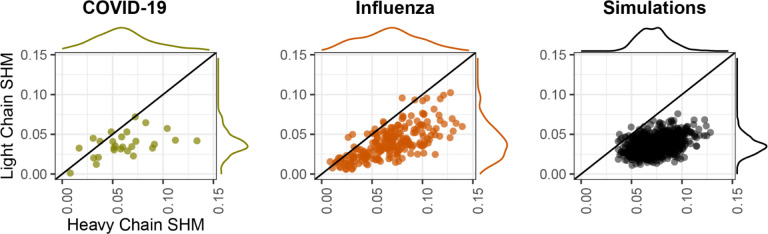
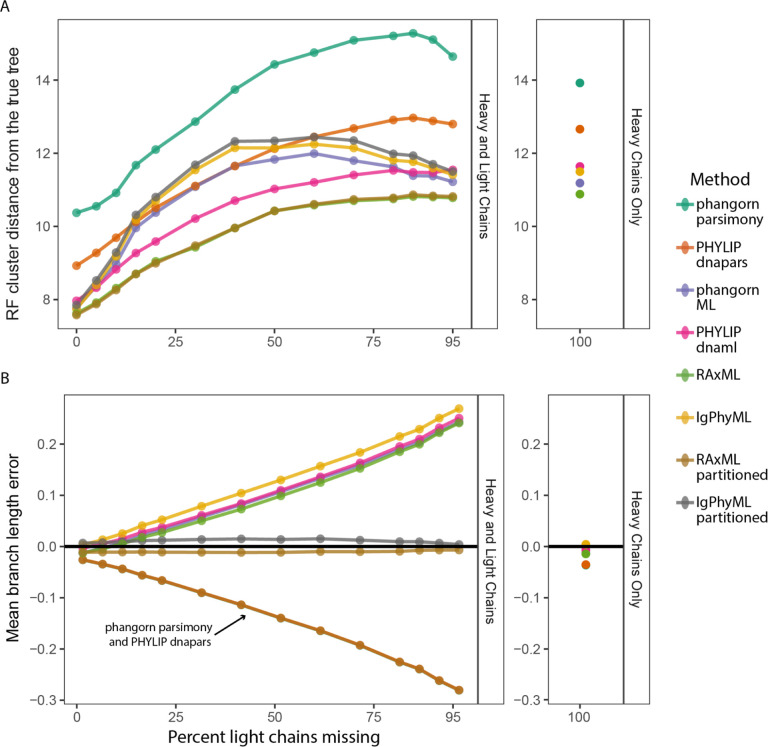
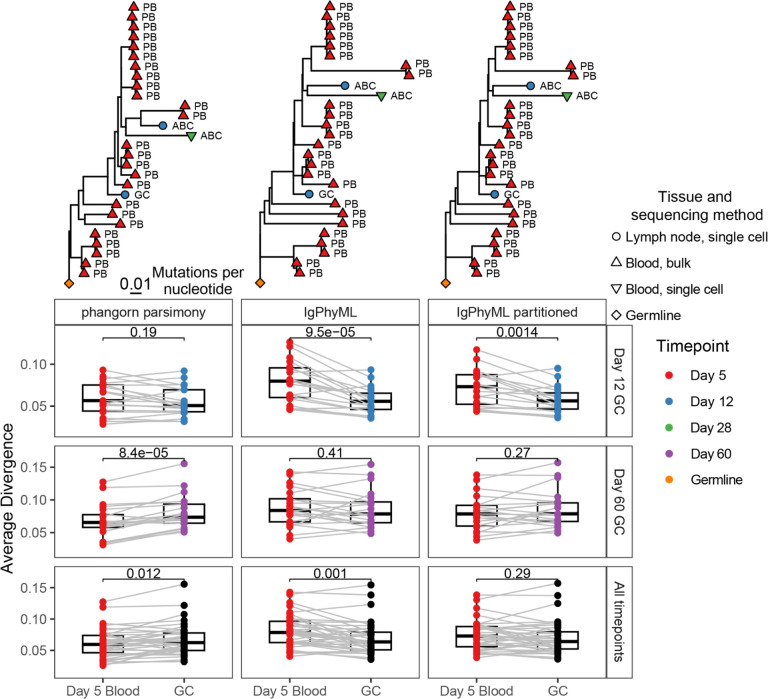
Antibodies are vital to human immune responses and are composed of genetically variable heavy and light chains. These structures are initially expressed as B cell receptors (BCRs). BCR diversity is shaped through somatic hypermutation and selection during immune responses. This evolutionary process produces B cell clones, cells that descend from a common ancestor but differ by mutations. Phylogenetic trees inferred from BCR sequences can reconstruct the history of mutations within a clone. Until recently, BCR sequencing technologies separated heavy and light chains, but advancements in single cell sequencing now pair heavy and light chains from individual cells. However, it is unclear how these separate genes should be combined to infer B cell phylogenies. In this study, we investigated strategies for using paired heavy and light chain sequences to build phylogenetic trees. We found incorporating light chains significantly improved tree accuracy and reproducibility across all methods tested. This improvement was greater than the difference between tree building methods and persisted even when mixing bulk and single cell sequencing data. However, we also found that many phylogenetic methods estimated significantly biased branch lengths when some light chains were missing, such as when mixing single cell and bulk BCR data. This bias was eliminated using maximum likelihood methods with separate branch lengths for heavy and light chain gene partitions. Thus, we recommend using maximum likelihood methods with separate heavy and light chain partitions, especially when mixing data types. We implemented these methods in the R package Dowser: https://dowser.readthedocs.io.
Conflict of interest statement
Competing interests: S.H.K. receives consulting fees from Peraton. K.B.H. receives consulting fees from Prellis Biologics. The remaining authors have no competing interests.
Figures


References
-
- Murphy K., Weaver C. & Berg L. Janeway’s Immunobiology. (2022).
-
- Victora G. D. & Nussenzweig M. C. Germinal Centers. Annu. Rev. Immunol. 30, 429–457 (2012). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources