

This is a preprint.
Initiation of a ZAKα-dependent Ribotoxic Stress Response by the Innate Immunity Endoribonuclease RNase L
- PMID: 37873202
- PMCID: PMC10592832
- DOI: 10.1101/2023.10.12.562082
Initiation of a ZAKα-dependent Ribotoxic Stress Response by the Innate Immunity Endoribonuclease RNase L
Update in
-
Initiation of a ZAKα-dependent ribotoxic stress response by the innate immunity endoribonuclease RNase L.Cell Rep. 2024 Apr 23;43(4):113998. doi: 10.1016/j.celrep.2024.113998. Epub 2024 Mar 28. Cell Rep. 2024. PMID: 38551960 Free PMC article.
Abstract
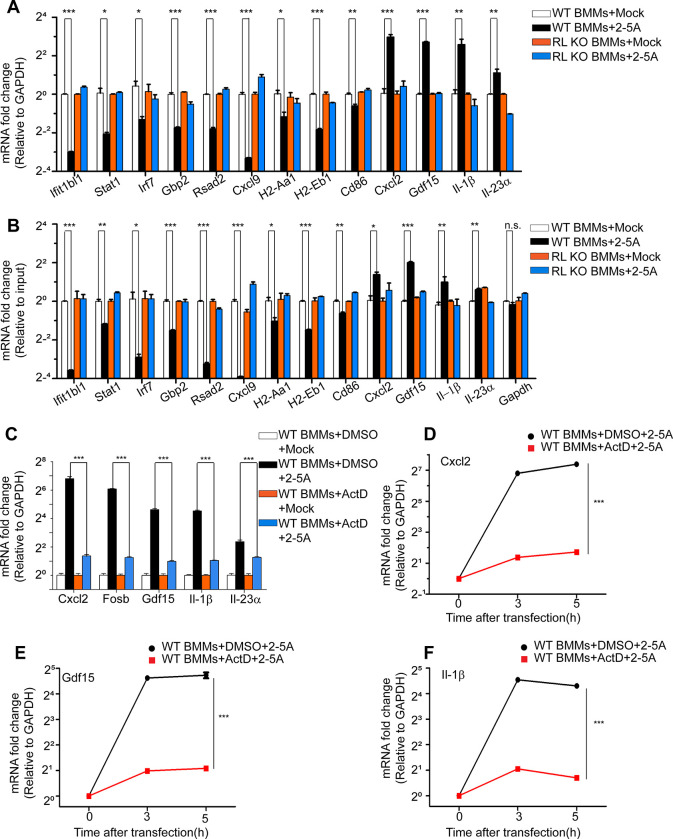
RNase L is a regulated endoribonuclease in higher vertebrates that functions in antiviral innate immunity. Interferons induce OAS enzymes that sense double-stranded RNA of viral origin leading to synthesis of 2',5'-oligoadenylate (2-5A) activators of RNase L. However, it is unknown precisely how RNase L inhibits viral infections. To isolate effects of RNase L from other effects of double-stranded RNA or virus, 2-5A was directly introduced into cells. Here we report that RNase L activation by 2-5A causes a ribotoxic stress response that requires the ribosome-associated MAP3K, ZAKα. Subsequently, the stress-activated protein kinases (SAPK) JNK and p38α are phosphorylated. RNase L activation profoundly altered the transcriptome by widespread depletion of mRNAs associated with different cellular functions, but also by SAPK-dependent induction of inflammatory genes. Our findings show that 2-5A is a ribotoxic stressor that causes RNA damage through RNase L triggering a ZAKα kinase cascade leading to proinflammatory signaling and apoptosis.
Keywords: 2–5A; Innate immunity; RNase L; ZAKα; ribotoxic stress response.
Figures







References
-
- Iordanov M.S., Pribnow D., Magun J.L., Dinh T.H., Pearson J.A., Chen S.L., and Magun B.E. (1997). Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 17, 3373–3381. - PMC - PubMed
-
- Vind A.C., Snieckute G., Blasius M., Tiedje C., Krogh N., Bekker-Jensen D.B., Andersen K.L., Nordgaard C., Tollenaere M.A.X., Lund A.H., et al. (2020). ZAKalpha Recognizes Stalled Ribosomes through Partially Redundant Sensor Domains. Mol Cell 78, 700–713 e707. 10.1016/j.molcel.2020.03.021. - DOI - PubMed
-
- Iordanov M.S., Pribnow D., Magun J.L., Dinh T.H., Pearson J.A., and Magun B.E. (1998). Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J Biol Chem 273, 15794–15803. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials