

This is a preprint.
Deep Mutational Scanning in Disease-related Genes with Saturation Mutagenesis-Reinforced Functional Assays (SMuRF)
- PMID: 37873263
- PMCID: PMC10592615
- DOI: 10.1101/2023.07.12.548370
Deep Mutational Scanning in Disease-related Genes with Saturation Mutagenesis-Reinforced Functional Assays (SMuRF)
Update in
-
Saturation mutagenesis-reinforced functional assays for disease-related genes.Cell. 2024 Nov 14;187(23):6707-6724.e22. doi: 10.1016/j.cell.2024.08.047. Epub 2024 Sep 25. Cell. 2024. PMID: 39326416
Abstract
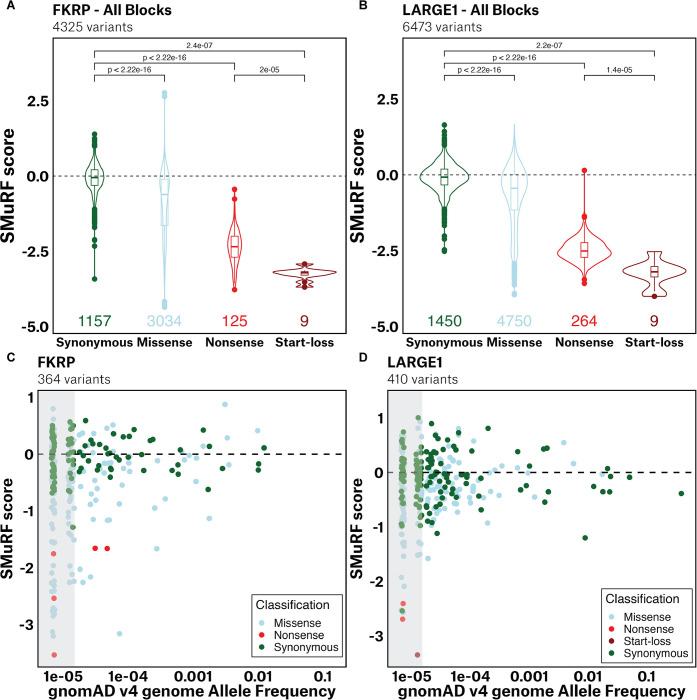
Interpretation of disease-causing genetic variants remains a challenge in human genetics. Current costs and complexity of deep mutational scanning methods hamper crowd-sourcing approaches toward genome-wide resolution of variants in disease-related genes. Our framework, Saturation Mutagenesis-Reinforced Functional assays (SMuRF), addresses these issues by offering simple and cost-effective saturation mutagenesis, as well as streamlining functional assays to enhance the interpretation of unresolved variants. Applying SMuRF to neuromuscular disease genes FKRP and LARGE1, we generated functional scores for all possible coding single nucleotide variants, which aid in resolving clinically reported variants of uncertain significance. SMuRF also demonstrates utility in predicting disease severity, resolving critical structural regions, and providing training datasets for the development of computational predictors. Our approach opens new directions for enabling variant-to-function insights for disease genes in a manner that is broadly useful for crowd-sourcing implementation across standard research laboratories.
Keywords: Deep mutational scanning; cost-effective variant interpretation; diagnostics; dystroglycanopathies; genetic diseases; high-throughput functional assays; muscular dystrophies; saturation mutagenesis; variant effect prediction; variants of uncertain significance.
Conflict of interest statement
Declaration of interests The authors declare no competing interests. Declaration of generative AI and AI-assisted technologies We used ChatGPT 3.5 and Gemini to improve the readability and language in this manuscript. The manuscript was first drafted by us and polished with the tools sentence-by-sentence where we deemed necessary. We then reviewed and finalized the text. We take full responsibility for the contents of this manuscript.
Figures





Similar articles
-
Saturation mutagenesis-reinforced functional assays for disease-related genes.Cell. 2024 Nov 14;187(23):6707-6724.e22. doi: 10.1016/j.cell.2024.08.047. Epub 2024 Sep 25. Cell. 2024. PMID: 39326416
-
Deep mutational scanning of proteins in mammalian cells.Cell Rep Methods. 2023 Nov 20;3(11):100641. doi: 10.1016/j.crmeth.2023.100641. Epub 2023 Nov 13. Cell Rep Methods. 2023. PMID: 37963462 Free PMC article. Review.
-
Integrating deep mutational scanning and low-throughput mutagenesis data to predict the impact of amino acid variants.Gigascience. 2022 Dec 28;12:giad073. doi: 10.1093/gigascience/giad073. Epub 2023 Sep 18. Gigascience. 2022. PMID: 37721410 Free PMC article.
-
Reducing uncertainty in genetic testing with Saturation Genome Editing.Med Genet. 2022 Nov 29;34(4):297-304. doi: 10.1515/medgen-2022-2159. eCollection 2022 Dec. Med Genet. 2022. PMID: 38836089 Free PMC article.
-
Multiplexed assays of variant effects contribute to a growing genotype-phenotype atlas.Hum Genet. 2018 Sep;137(9):665-678. doi: 10.1007/s00439-018-1916-x. Epub 2018 Aug 2. Hum Genet. 2018. PMID: 30073413 Free PMC article. Review.
References
-
- Fridman H., Yntema H.G., Mägi R., Andreson R., Metspalu A., Mezzavila M., Tyler-Smith C., Xue Y., Carmi S., Levy-Lahad E., et al. (2021). The landscape of autosomal-recessive pathogenic variants in European populations reveals phenotype-specific effects. Am. J. Hum. Genet. 108, 608–619. 10.1016/j.ajhg.2021.03.004. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources