

This is a preprint.
TMEM65 regulates NCLX-dependent mitochondrial calcium efflux
- PMID: 37873405
- PMCID: PMC10592617
- DOI: 10.1101/2023.10.06.561062
TMEM65 regulates NCLX-dependent mitochondrial calcium efflux
Update in
-
TMEM65 regulates and is required for NCLX-dependent mitochondrial calcium efflux.Nat Metab. 2025 Apr;7(4):714-729. doi: 10.1038/s42255-025-01250-9. Epub 2025 Apr 8. Nat Metab. 2025. PMID: 40200126 Free PMC article.
Abstract
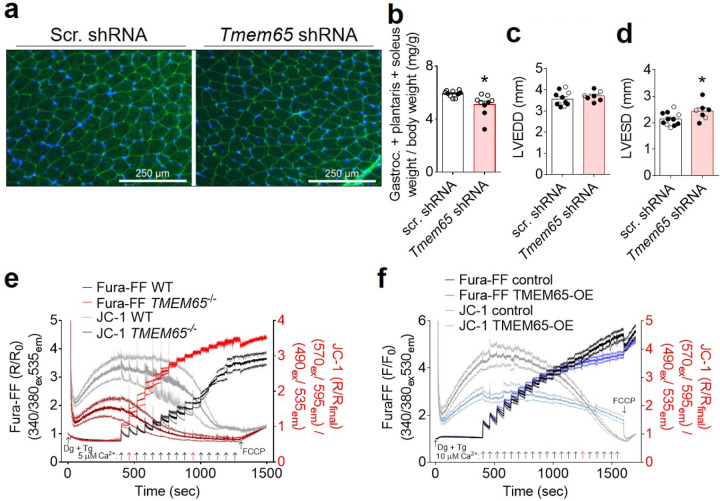
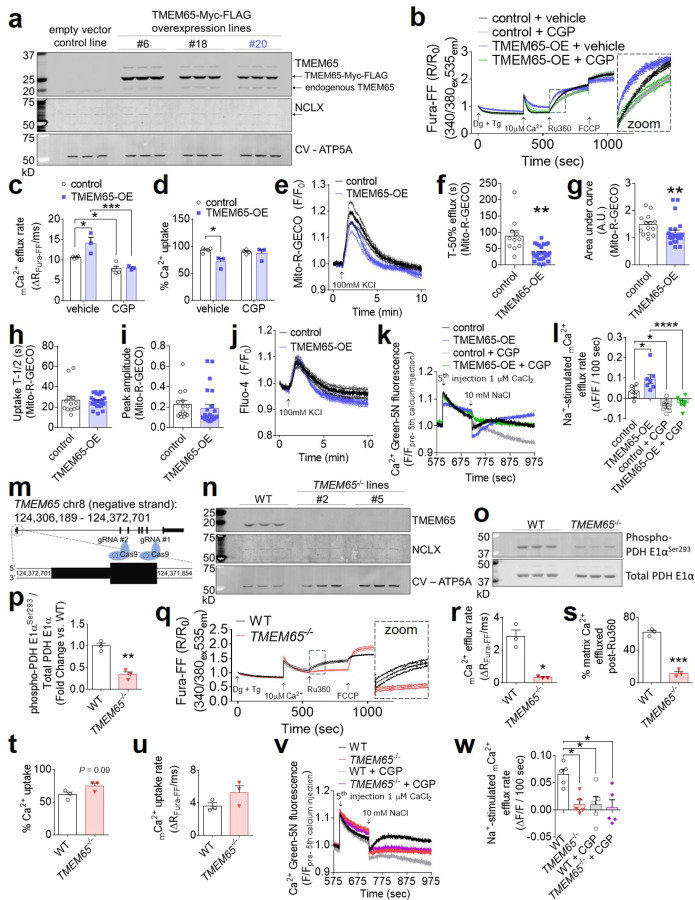
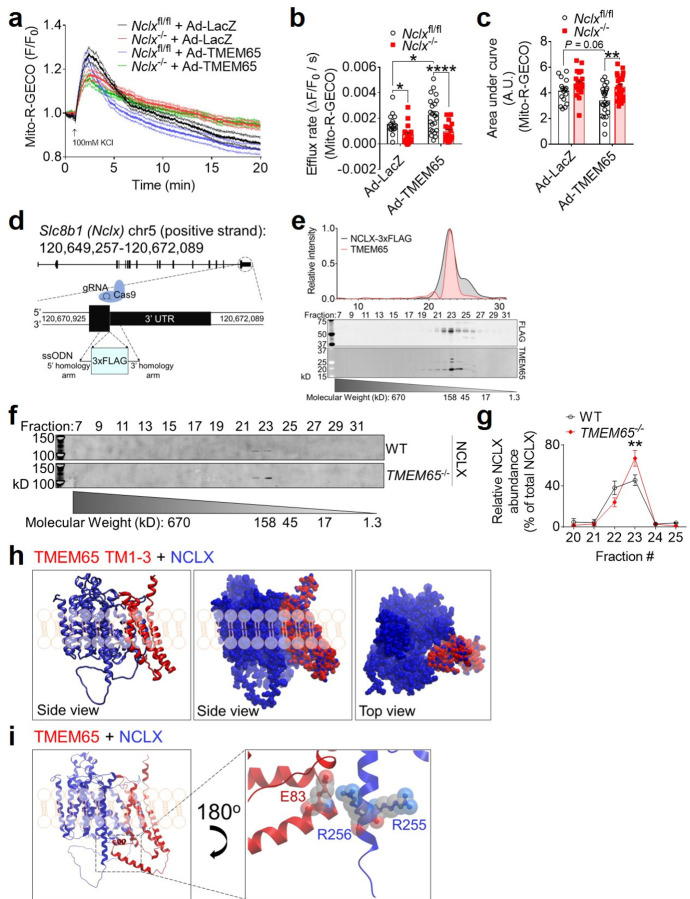
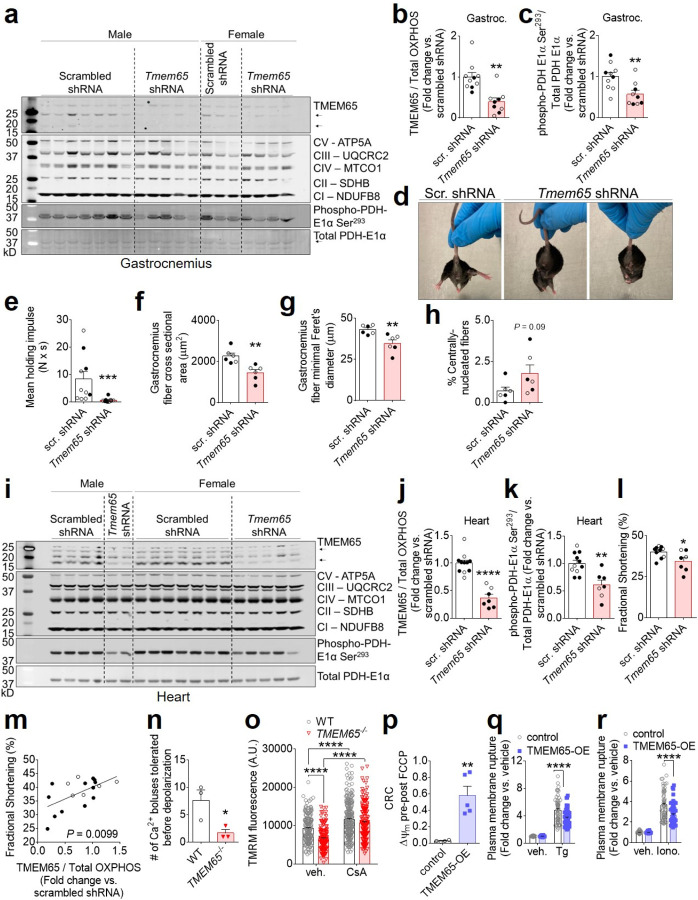
The balance between mitochondrial calcium (mCa2+) uptake and efflux regulates ATP production, but if perturbed causes energy starvation or mCa2+ overload and cell death. The mitochondrial sodium-calcium exchanger, NCLX, is a critical route of mCa2+ efflux in excitable tissues, such as the heart and brain, and animal models support NCLX as a promising therapeutic target to limit pathogenic mCa2+ overload. However, the mechanisms that regulate NCLX activity remain largely unknown. We used proximity biotinylation proteomic screening to identify the NCLX interactome and define novel regulators of NCLX function. Here, we discover the mitochondrial inner membrane protein, TMEM65, as an NCLX-proximal protein that potently enhances sodium (Na+)-dependent mCa2+ efflux. Mechanistically, acute pharmacologic NCLX inhibition or genetic deletion of NCLX ablates the TMEM65-dependent increase in mCa2+ efflux. Further, loss-of-function studies show that TMEM65 is required for Na+-dependent mCa2+ efflux. Co-fractionation and in silico structural modeling of TMEM65 and NCLX suggest these two proteins exist in a common macromolecular complex in which TMEM65 directly stimulates NCLX function. In line with these findings, knockdown of Tmem65 in mice promotes mCa2+ overload in the heart and skeletal muscle and impairs both cardiac and neuromuscular function. We further demonstrate that TMEM65 deletion causes excessive mitochondrial permeability transition, whereas TMEM65 overexpression protects against necrotic cell death during cellular Ca2+ stress. Collectively, our results show that loss of TMEM65 function in excitable tissue disrupts NCLX-dependent mCa2+ efflux, causing pathogenic mCa2+ overload, cell death and organ-level dysfunction, and that gain of TMEM65 function mitigates these effects. These findings demonstrate the essential role of TMEM65 in regulating NCLX-dependent mCa2+ efflux and suggest modulation of TMEM65 as a novel strategy for the therapeutic control of mCa2+ homeostasis.
Keywords: NCLX; TMEM65; calcium; mitochondria; sodium.
Conflict of interest statement
Competing Interests: None
Figures




References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous