

PLK3 facilitates replication of swine influenza virus by phosphorylating viral NP protein
- PMID: 37874309
- PMCID: PMC10768867
- DOI: 10.1080/22221751.2023.2275606
PLK3 facilitates replication of swine influenza virus by phosphorylating viral NP protein
Abstract
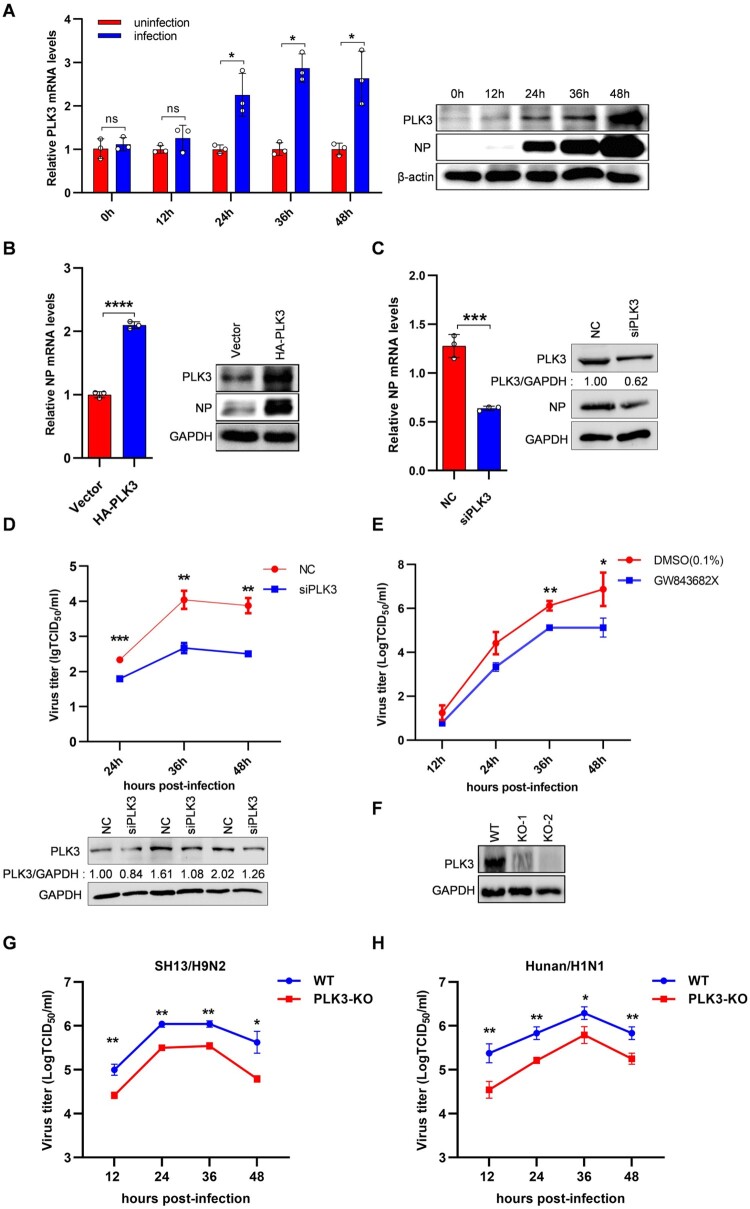
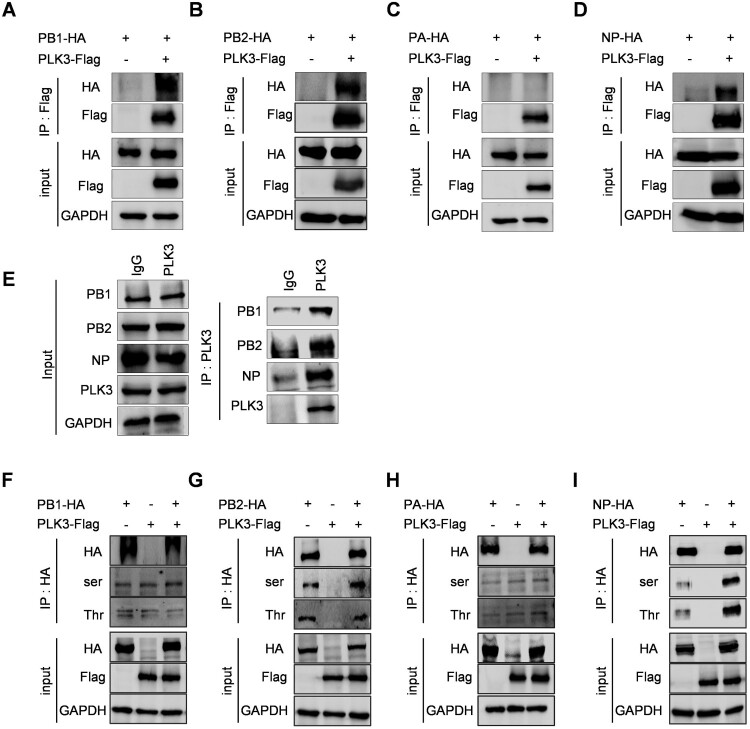
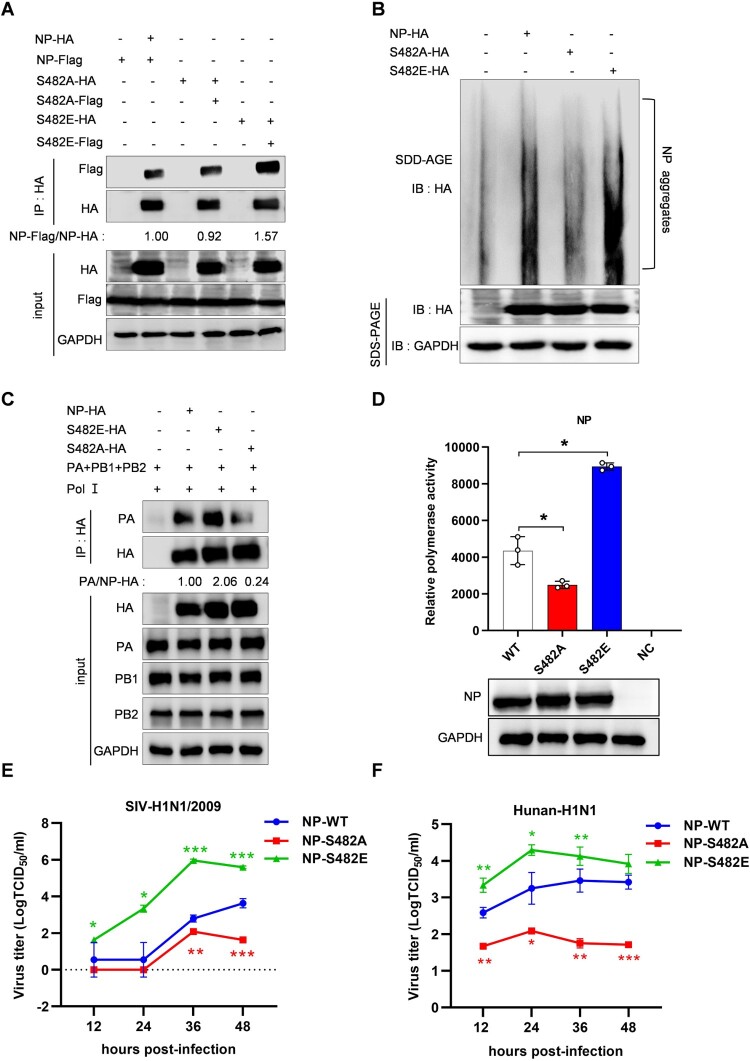
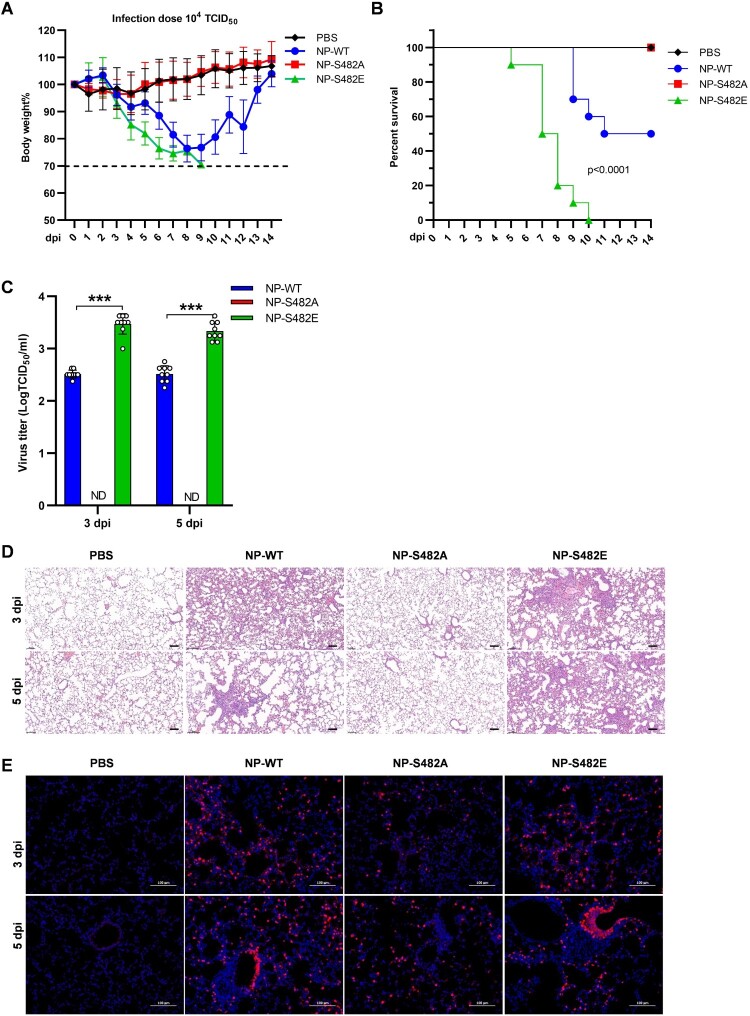
Swine H1N1/2009 influenza is a highly infectious respiratory disease in pigs, which poses a great threat to pig production and human health. In this study, we investigated the global expression profiling of swine-encoded genes in response to swine H1N1/2009 influenza A virus (SIV-H1N1/2009) in newborn pig trachea (NPTr) cells. In total, 166 genes were found to be differentially expressed (DE) according to the gene microarray. After analyzing the DE genes which might affect the SIV-H1N1/2009 replication, we focused on polo-like kinase 3 (PLK3). PLK3 is a member of the PLK family, which is a highly conserved serine/threonine kinase in eukaryotes and well known for its role in the regulation of cell cycle and cell division. We validated that the expression of PLK3 was upregulated after SIV-H1N1/2009 infection. Additionally, PLK3 was found to interact with viral nucleoprotein (NP), significantly increased NP phosphorylation and oligomerization, and promoted viral ribonucleoprotein assembly and replication. Furthermore, we identified serine 482 (S482) as the phosphorylated residue on NP by PLK3. The phosphorylation of S482 regulated NP oligomerization, viral polymerase activity and growth. Our findings provide further insights for understanding the replication of influenza A virus.
Keywords: PLK3; Swine influenza virus; nucleoprotein; phosphorylation; virus replication.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous