

Cell State of Origin Impacts Development of Distinct Endometriosis-Related Ovarian Carcinoma Histotypes
- PMID: 37874327
- PMCID: PMC10758692
- DOI: 10.1158/0008-5472.CAN-23-1362
Cell State of Origin Impacts Development of Distinct Endometriosis-Related Ovarian Carcinoma Histotypes
Abstract
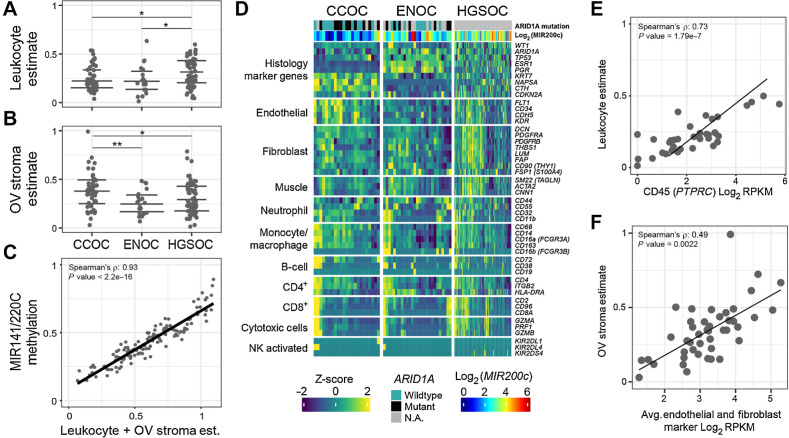
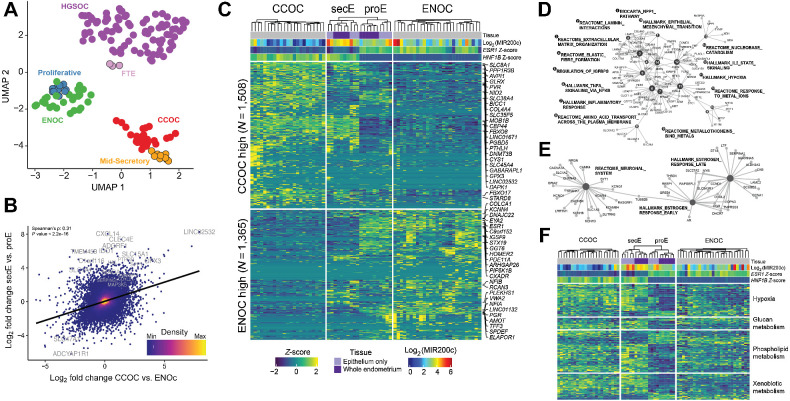
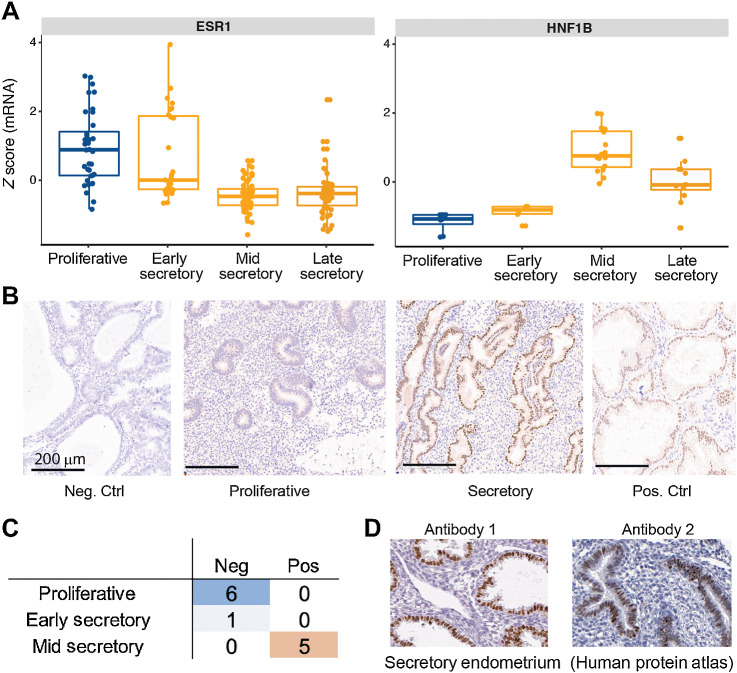
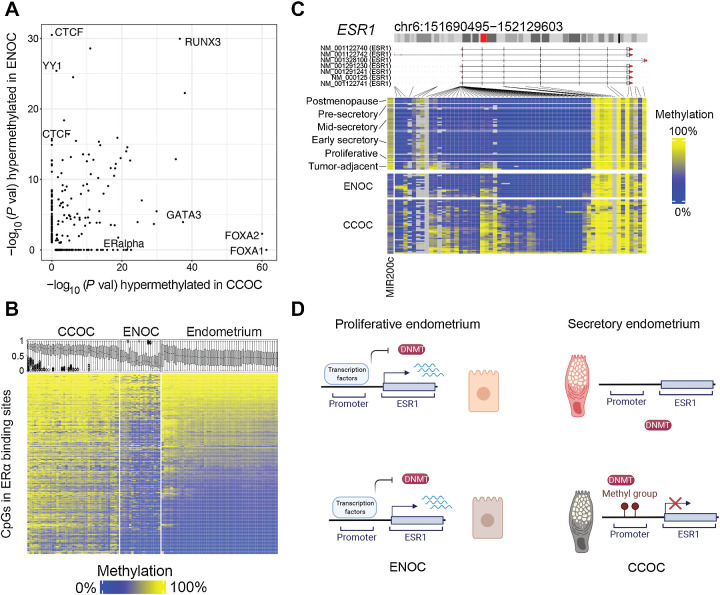
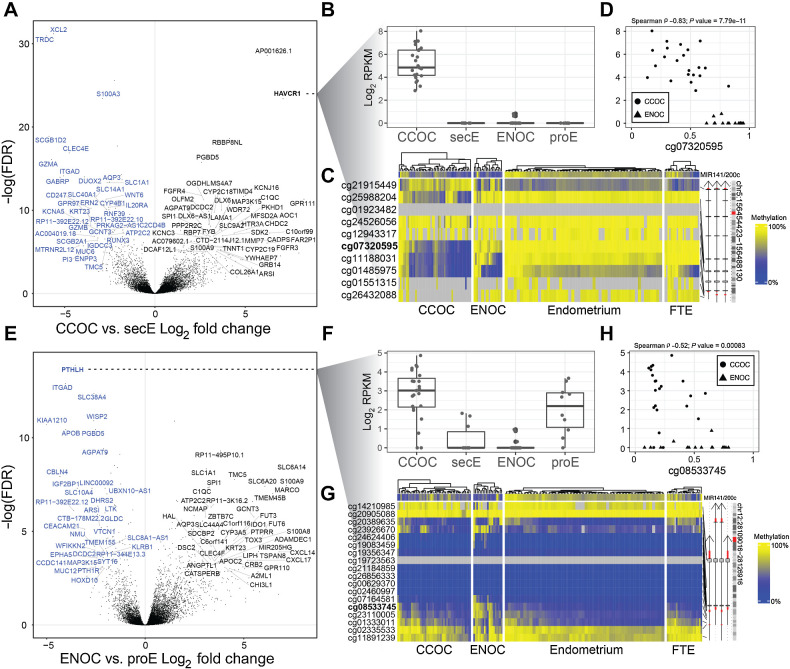
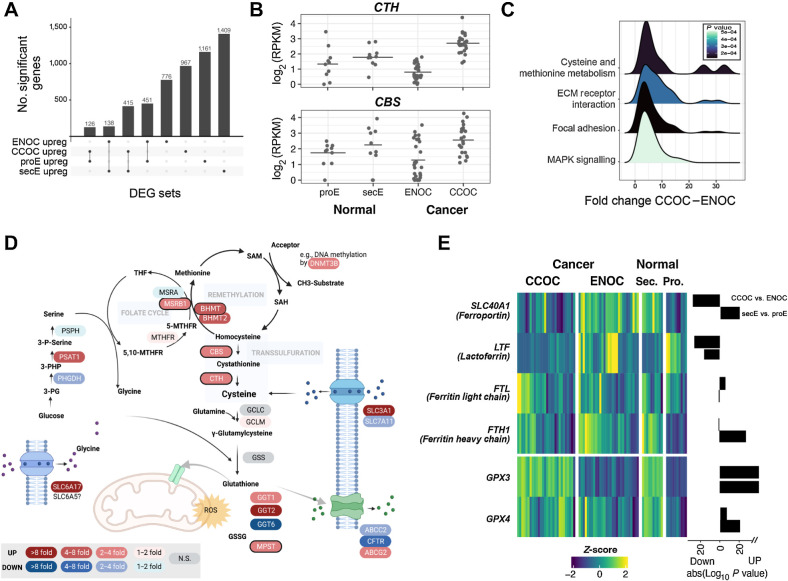
Clear cell ovarian carcinoma (CCOC) and endometrioid ovarian carcinoma (ENOC) are ovarian carcinoma histotypes, which are both thought to arise from ectopic endometrial (or endometrial-like) cells through an endometriosis intermediate. How the same cell type of origin gives rise to two morphologically and biologically different histotypes has been perplexing, particularly given that recurrent genetic mutations are common to both and present in nonmalignant precursors. We used RNA transcription analysis to show that the expression profiles of CCOC and ENOC resemble those of normal endometrium at secretory and proliferative phases of the menstrual cycle, respectively. DNA methylation at the promoter of the estrogen receptor (ER) gene (ESR1) was enriched in CCOC, which could potentially lock the cells in the secretory state. Compared with normal secretory-type endometrium, CCOC was further defined by increased expression of cysteine and glutathione synthesis pathway genes and downregulation of the iron antiporter, suggesting iron addiction and highlighting ferroptosis as a potential therapeutic target. Overall, these findings suggest that while CCOC and ENOC arise from the same cell type, these histotypes likely originate from different cell states. This "cell state of origin" model may help to explain the presence of histologic and molecular cancer subtypes arising in other organs.
Significance: Two cancer histotypes diverge from a common cell of origin epigenetically locked in different cell states, highlighting the importance of considering cell state to better understand the cell of origin of cancer.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
