

Radical and Cationic Pathways in C(sp3)-H Bond Oxygenation by Dioxiranes of Bicyclic and Spirocyclic Hydrocarbons Bearing Cyclopropane Moieties
- PMID: 37874906
- PMCID: PMC10636757
- DOI: 10.1021/jacs.3c07163
Radical and Cationic Pathways in C(sp3)-H Bond Oxygenation by Dioxiranes of Bicyclic and Spirocyclic Hydrocarbons Bearing Cyclopropane Moieties
Abstract
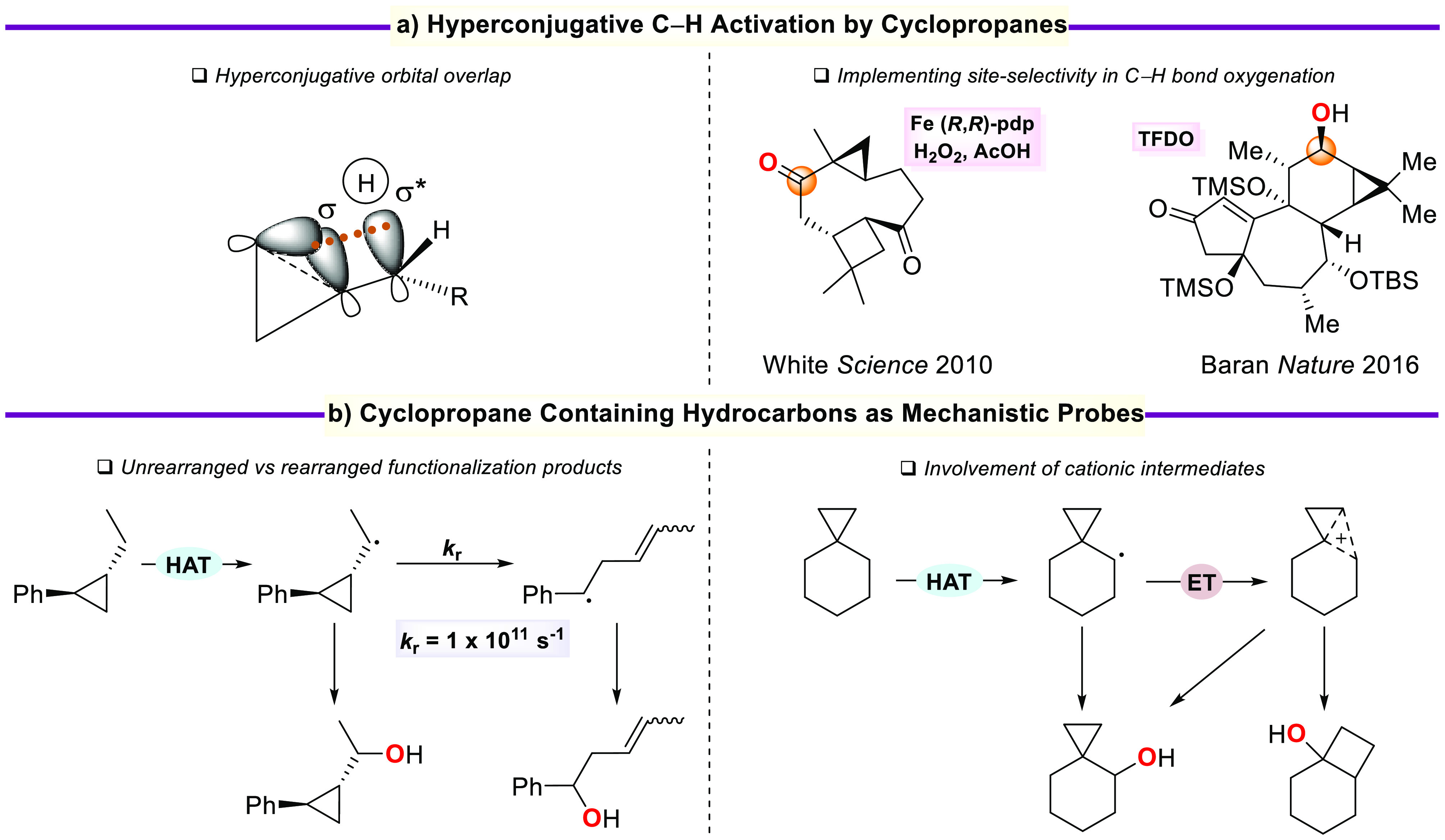
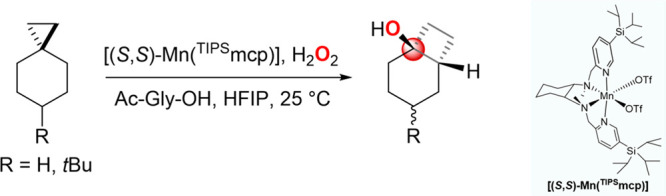
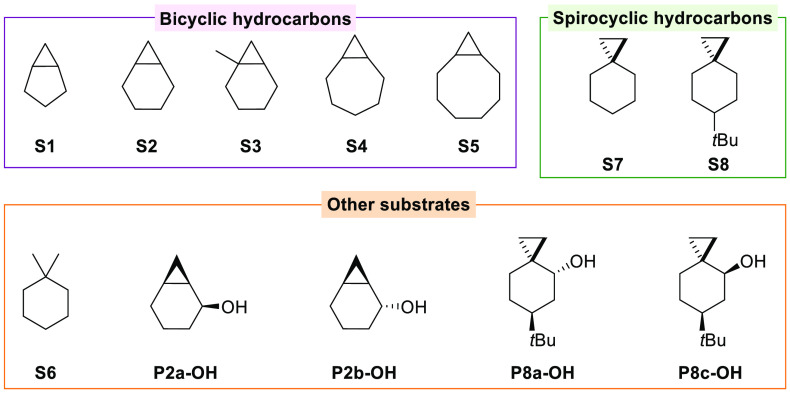
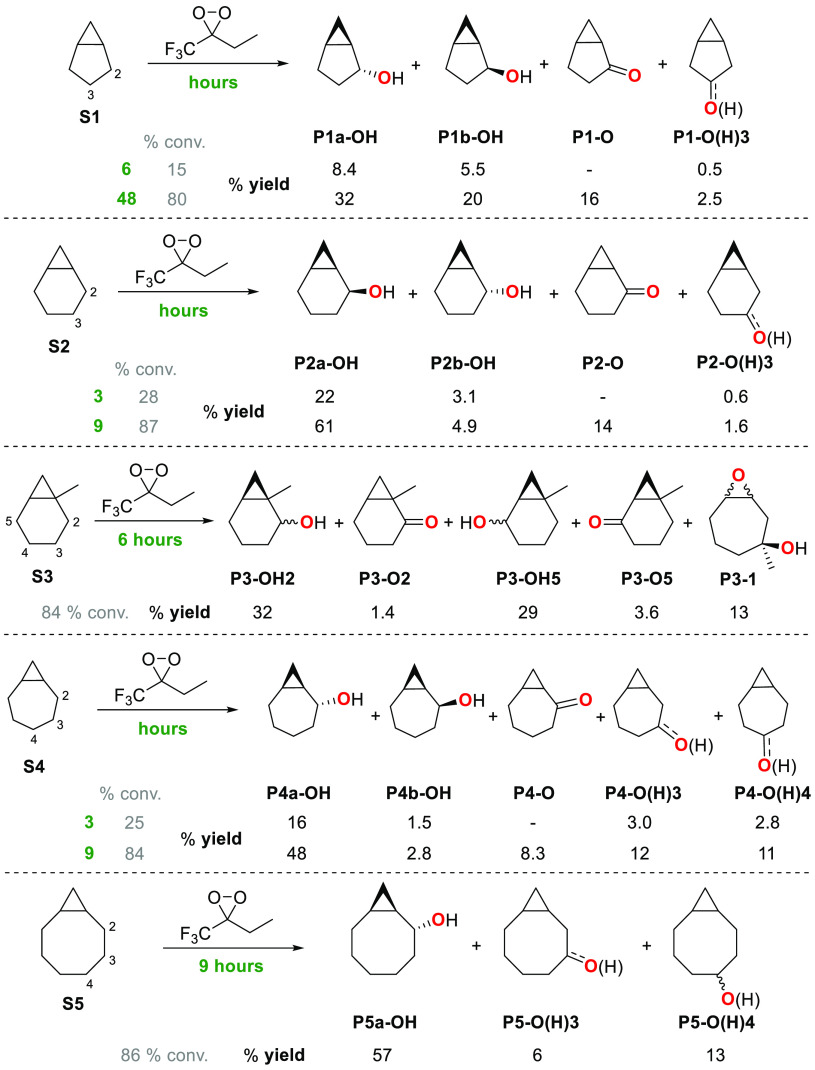
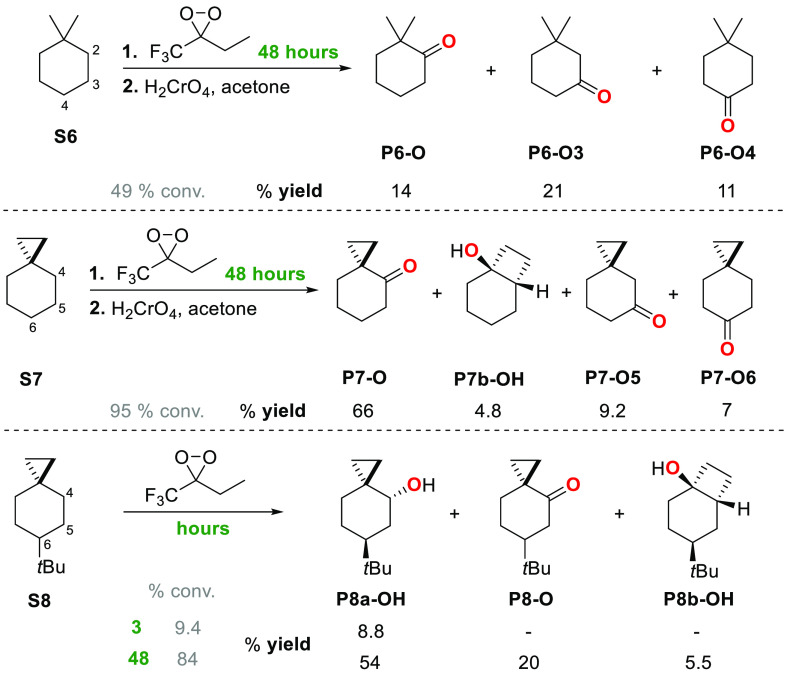
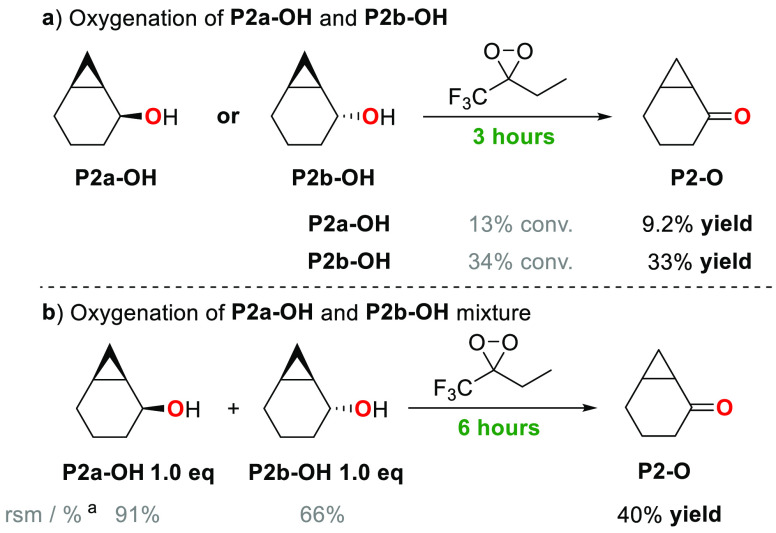
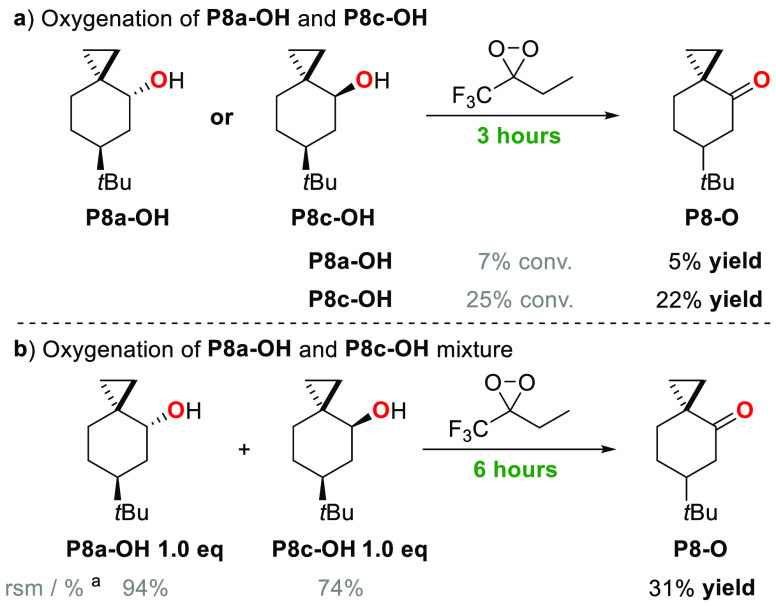
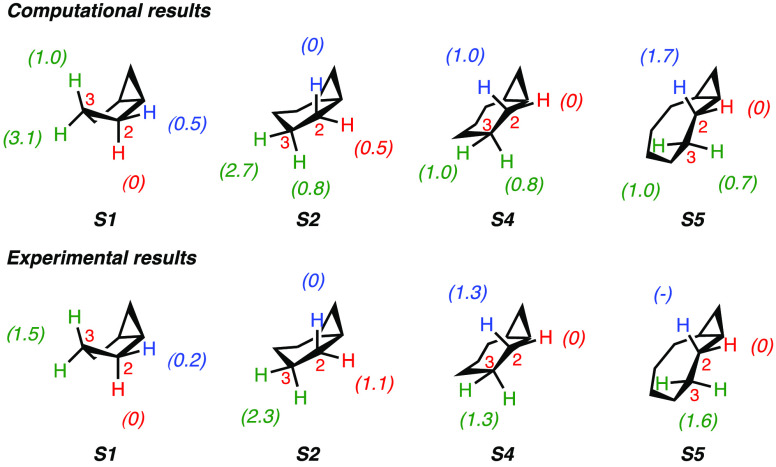
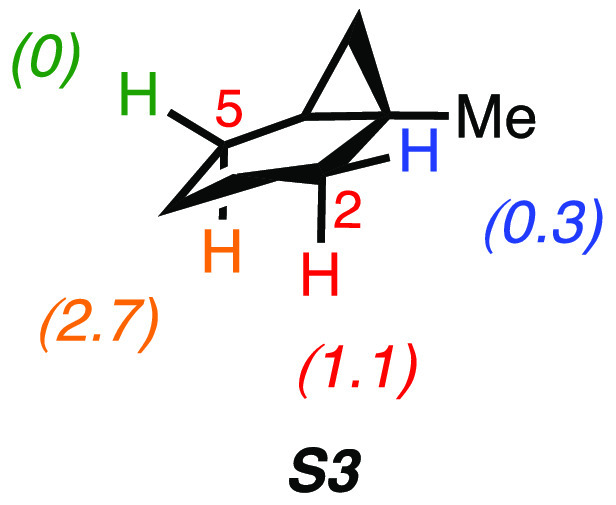
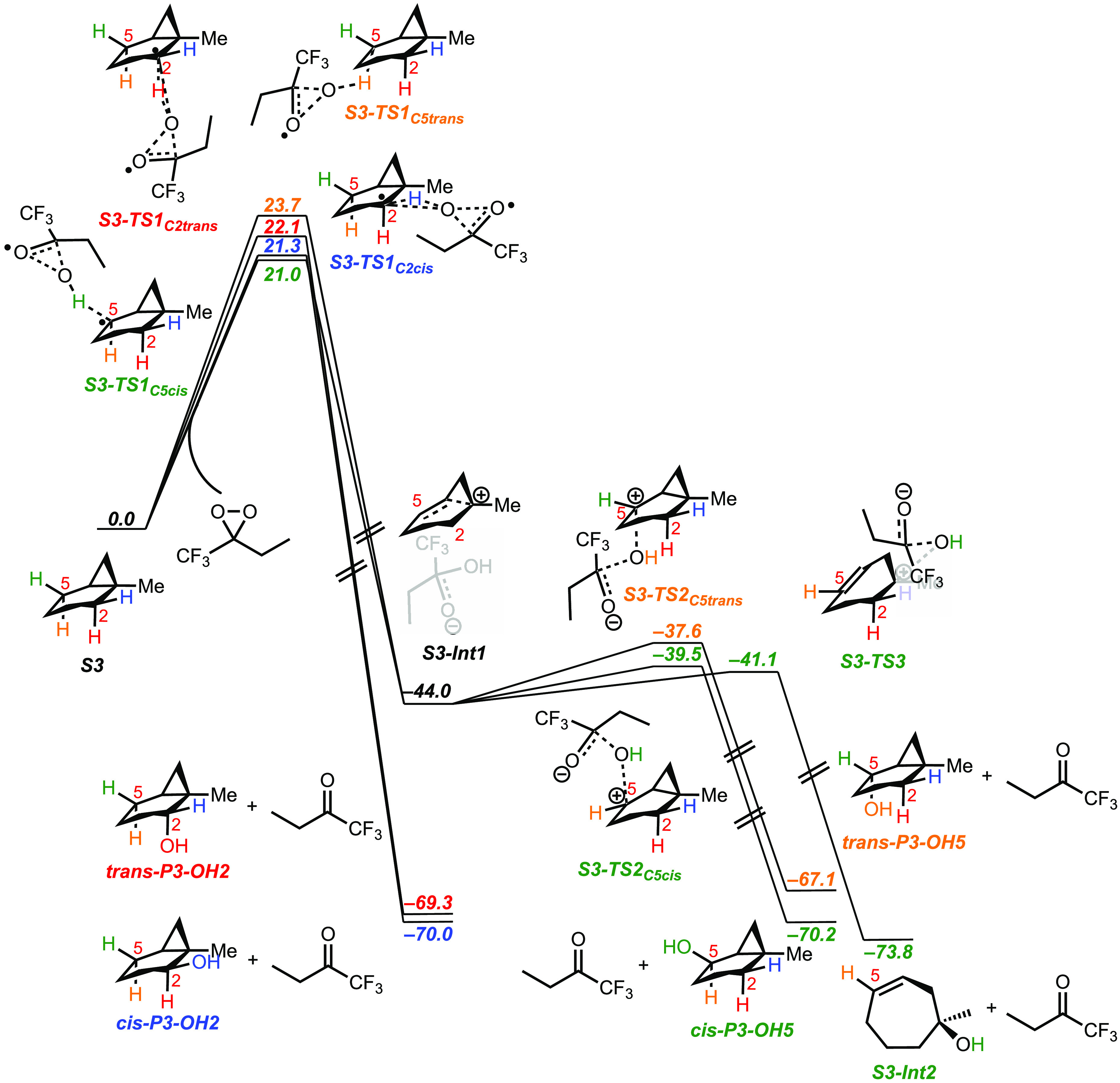
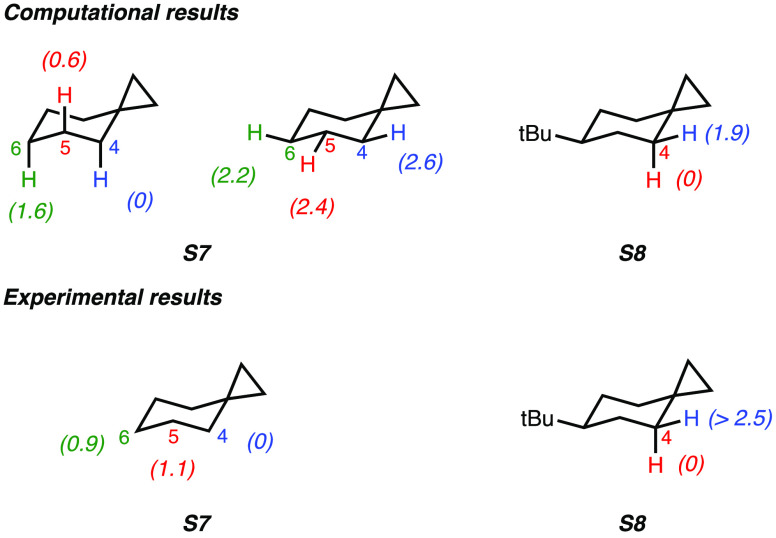
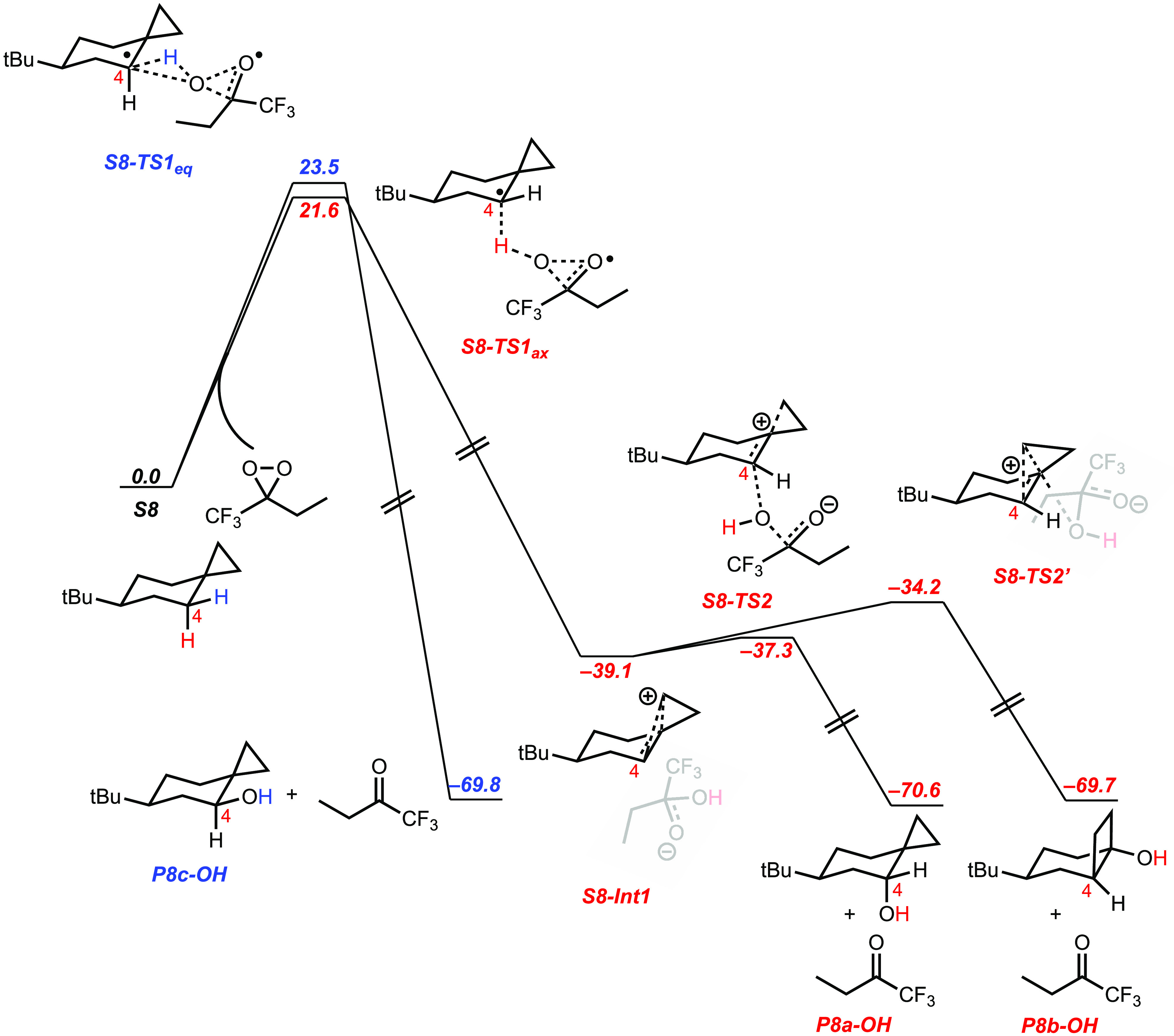
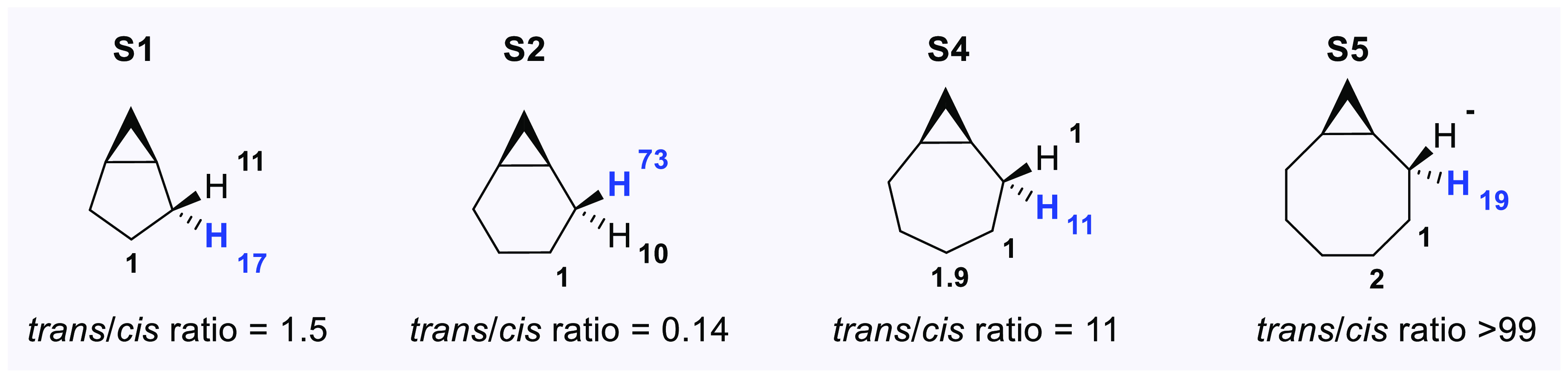
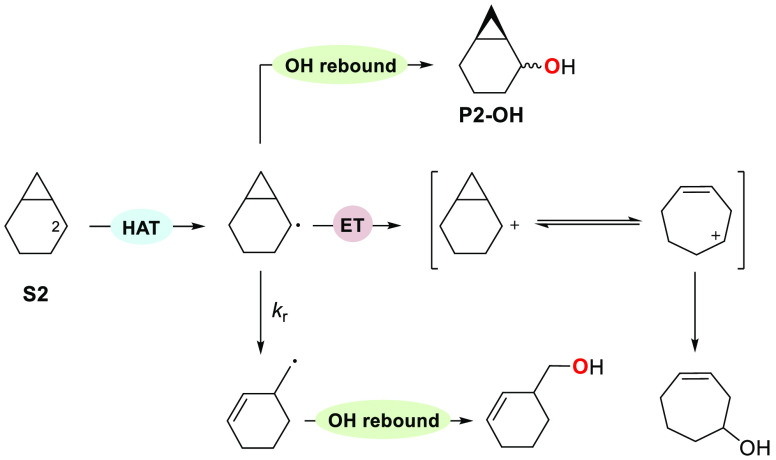
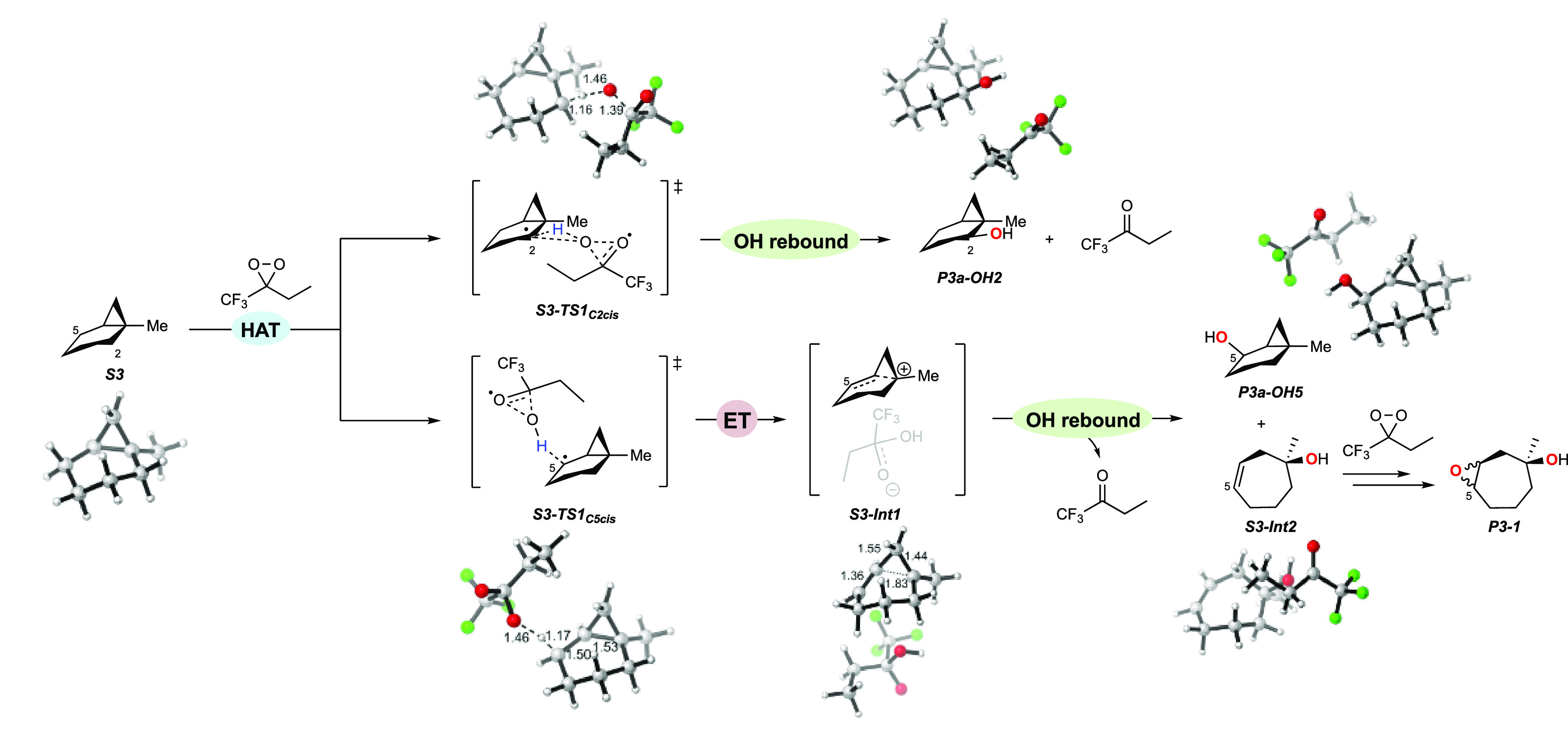
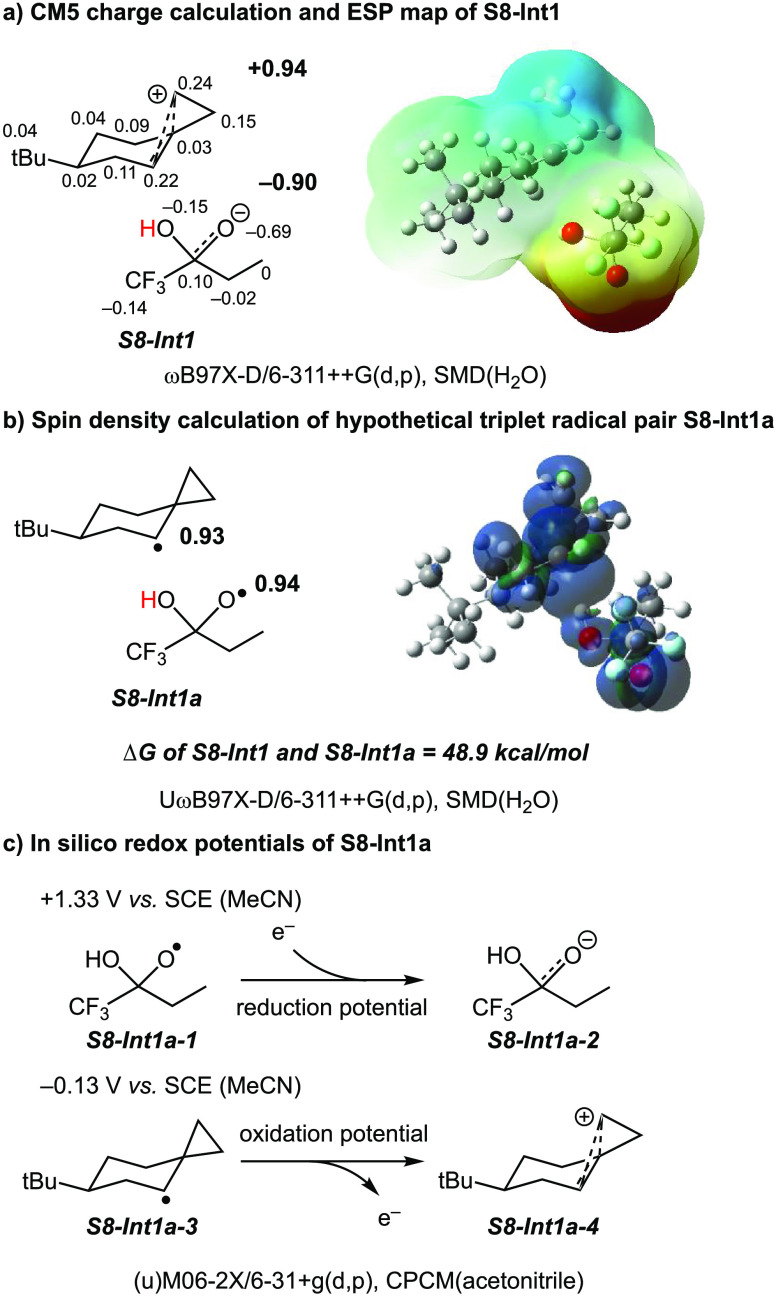
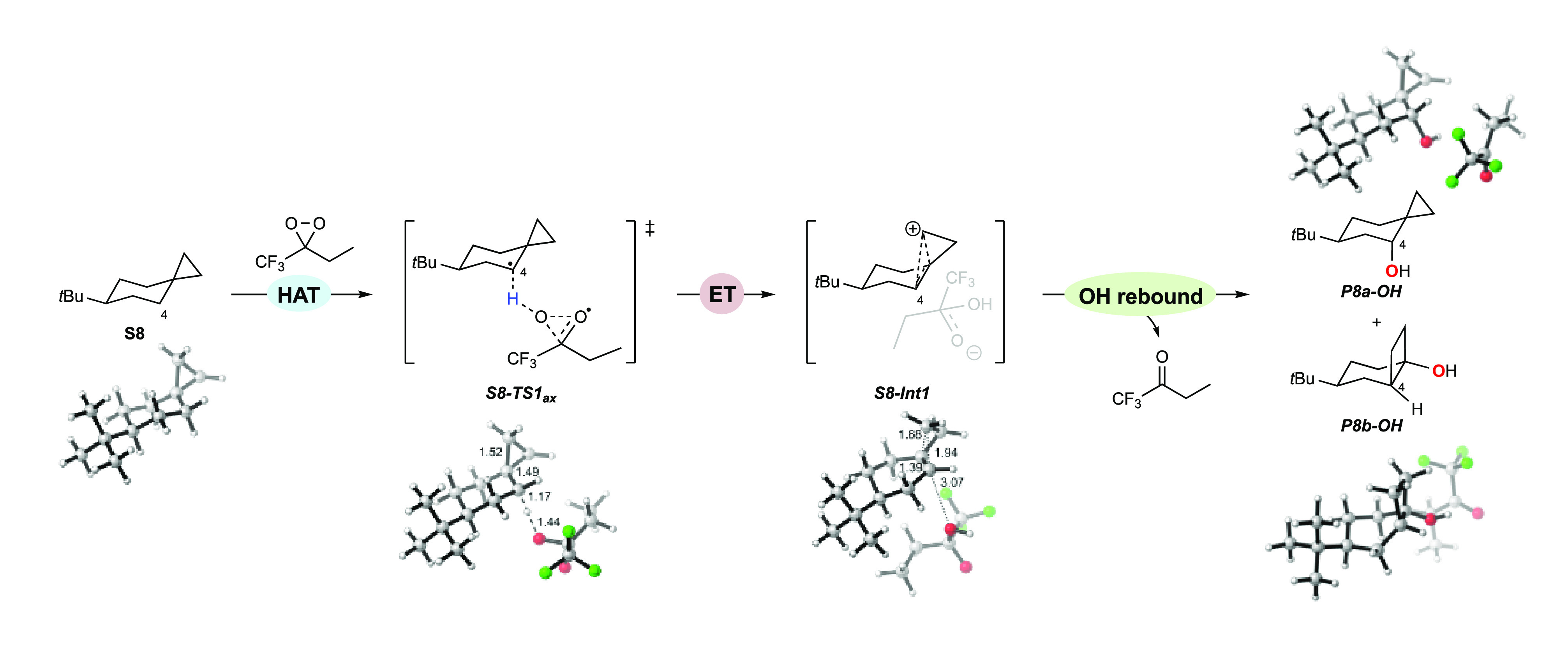
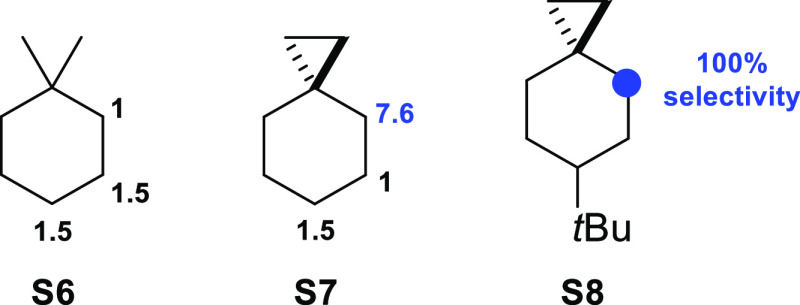
A product and DFT computational study on the reactions of 3-ethyl-3-(trifluoromethyl)dioxirane (ETFDO) with bicyclic and spirocyclic hydrocarbons bearing cyclopropyl groups was carried out. With bicyclo[n.1.0]alkanes (n = 3-6), diastereoselective formation of the alcohol product derived from C2-H bond hydroxylation was observed, accompanied by smaller amounts of products derived from oxygenation at other sites. With 1-methylbicyclo[4.1.0]heptane, rearranged products were also observed in addition to the unrearranged products deriving from oxygenation at the most activated C2-H and C5-H bonds. With spiro[2.5]octane and 6-tert-butylspiro[2.5]octane, reaction with ETFDO occurred predominantly or exclusively at the axial C4-H to give unrearranged oxygenation products, accompanied by smaller amounts of rearranged bicyclo[4.2.0]octan-1-ols. The good to outstanding site-selectivities and diastereoselectivities are paralleled by the calculated activation free energies for the corresponding reaction pathways. Computations show that the σ* orbitals of the bicyclo[n.1.0]alkane cis or trans C2-H bonds and spiro[2.5]octanes axial C4-H bond hyperconjugatively interact with the Walsh orbitals of the cyclopropane ring, activating these bonds toward HAT to ETFDO. The detection of rearranged oxygenation products in the oxidation of 1-methylbicyclo[4.1.0]heptane, spiro[2.5]octane, and 6-tert-butylspiro[2.5]octane provides unambiguous evidence for the involvement of cationic intermediates in these reactions, representing the first examples on the operation of ET pathways in dioxirane-mediated C(sp3)-H bond oxygenations. Computations support these findings, showing that formation of cationic intermediates is associated with specific stabilizing hyperconjugative interactions between the incipient carbon radical and the cyclopropane C-C bonding orbitals that trigger ET to the incipient dioxirane derived 1,1,1-trifluoro-2-hydroxy-2-butoxyl radical.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

Similar articles
-
Resolving Oxygenation Pathways in Manganese-Catalyzed C(sp3)-H Functionalization via Radical and Cationic Intermediates.J Am Chem Soc. 2022 Apr 27;144(16):7391-7401. doi: 10.1021/jacs.2c01466. Epub 2022 Apr 13. J Am Chem Soc. 2022. PMID: 35417154 Free PMC article.
-
Oxyfunctionalization of non-natural targets by dioxiranes. 5. Selective oxidation of hydrocarbons bearing cyclopropyl moieties.J Org Chem. 2003 Oct 3;68(20):7806-10. doi: 10.1021/jo034768o. J Org Chem. 2003. PMID: 14510559
-
Catalyst and Medium Control over Rebound Pathways in Manganese-Catalyzed Methylenic C-H Bond Oxidation.J Am Chem Soc. 2024 Apr 3;146(13):8904-8914. doi: 10.1021/jacs.3c11555. Epub 2024 Mar 20. J Am Chem Soc. 2024. PMID: 38506665 Free PMC article.
-
Continued Progress towards Efficient Functionalization of Natural and Non-natural Targets under Mild Conditions: Oxygenation by C-H Bond Activation with Dioxirane.Chemistry. 2019 Sep 18;25(52):12003-12017. doi: 10.1002/chem.201901687. Epub 2019 Jul 28. Chemistry. 2019. PMID: 31150563 Review.
-
Metabolism of Hydrocarbons in n-Alkane-Utilizing Anaerobic Bacteria.J Mol Microbiol Biotechnol. 2016;26(1-3):138-51. doi: 10.1159/000442160. Epub 2016 Mar 10. J Mol Microbiol Biotechnol. 2016. PMID: 26959725 Review.
Cited by
-
Biomimetic Frustrated Lewis Pair Catalysts for Hydrogenation of CO to Methanol at Low Temperatures.ACS Org Inorg Au. 2024 Jan 31;4(2):258-267. doi: 10.1021/acsorginorgau.3c00064. eCollection 2024 Apr 3. ACS Org Inorg Au. 2024. PMID: 38585511 Free PMC article.
-
Shaping of a Reactive Manganese Catalyst Enables Access to Polyfunctionalized Cyclohexanes via Enantioselective C(sp3)─H Bond Oxidation of 1,3-meso Diethers.Angew Chem Int Ed Engl. 2025 Jul 21;64(30):e202507755. doi: 10.1002/anie.202507755. Epub 2025 Jun 1. Angew Chem Int Ed Engl. 2025. PMID: 40392708 Free PMC article.
References
-
- De Meijere A. Bonding Properties of Cyclopropane and Their Chemical Consequences. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. 10.1002/anie.197908093. - DOI
-
- Schneider T. F.; Kaschel J.; Werz D. B. A New Golden Age for Donor–Acceptor Cyclopropanes. Angew. Chem. Int. Ed. 2014, 53, 5504–5523. 10.1002/anie.201309886. - DOI - PubMed
- Cohen Y.; Cohen A.; Marek I. Creating Stereocenters within Acyclic Systems by C–C Bond Cleavage of Cyclopropanes. Chem. Rev. 2021, 121, 140–161. 10.1021/acs.chemrev.0c00167. - DOI - PubMed
- Pirenne V.; Muriel B.; Waser J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2021, 121, 227–263. 10.1021/acs.chemrev.0c00109. - DOI - PubMed
-
- de Meijere A.; Kozhushkov S. I.; Schill H. Three-Membered-Ring-Based Molecular Architectures. Chem. Rev. 2006, 106, 4926–4996. 10.1021/cr0505369. - DOI - PubMed
- Mizuno A.; Matsui K.; Shuto S. From Peptides to Peptidomimetics: A Strategy Based on the Structural Features of Cyclopropane. Chem.—Eur. J. 2017, 23, 14394–14409. 10.1002/chem.201702119. - DOI - PubMed
-
- Wessjohann L. A.; Brandt W.; Thiemann T. Biosynthesis and Metabolism of Cyclopropane Rings in Natural Compounds. Chem. Rev. 2003, 103, 1625–1648. 10.1021/cr0100188. - DOI - PubMed
- Chen D. Y.-K.; Pouwer R. H.; Richard J.-A. Recent advances in the total synthesis of cyclopropane-containing natural products. Chem. Soc. Rev. 2012, 41, 4631–4642. 10.1039/c2cs35067j. - DOI - PubMed
- Fan Y.-Y.; Gao X.-H.; Yue J.-M. Attractive natural products with strained cyclopropane and/or cyclobutane ring systems. Sci. China Chem. 2016, 59, 1126–1141. 10.1007/s11426-016-0233-1. - DOI
- Brill Z. G.; Condakes M. L.; Ting C. P.; Maimone T. J. Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev. 2017, 117, 11753–11795. 10.1021/acs.chemrev.6b00834. - DOI - PMC - PubMed
- Li X.; Shimaya R.; Dairi T.; Chang W.-C.; Ogasawara Y. Identification of Cyclopropane Formation in the Biosyntheses of Hormaomycins and Belactosins: Sequential Nitration and Cyclopropanation by Metalloenzymes. Angew. Chem. Int. Ed. 2022, 61, e20211318910.1002/anie.202113189. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
