

Loss-of-Function but Not Gain-of-Function Properties of Mutant TP53 Are Critical for the Proliferation, Survival, and Metastasis of a Broad Range of Cancer Cells
- PMID: 37877779
- PMCID: PMC10850947
- DOI: 10.1158/2159-8290.CD-23-0402
Loss-of-Function but Not Gain-of-Function Properties of Mutant TP53 Are Critical for the Proliferation, Survival, and Metastasis of a Broad Range of Cancer Cells
Abstract
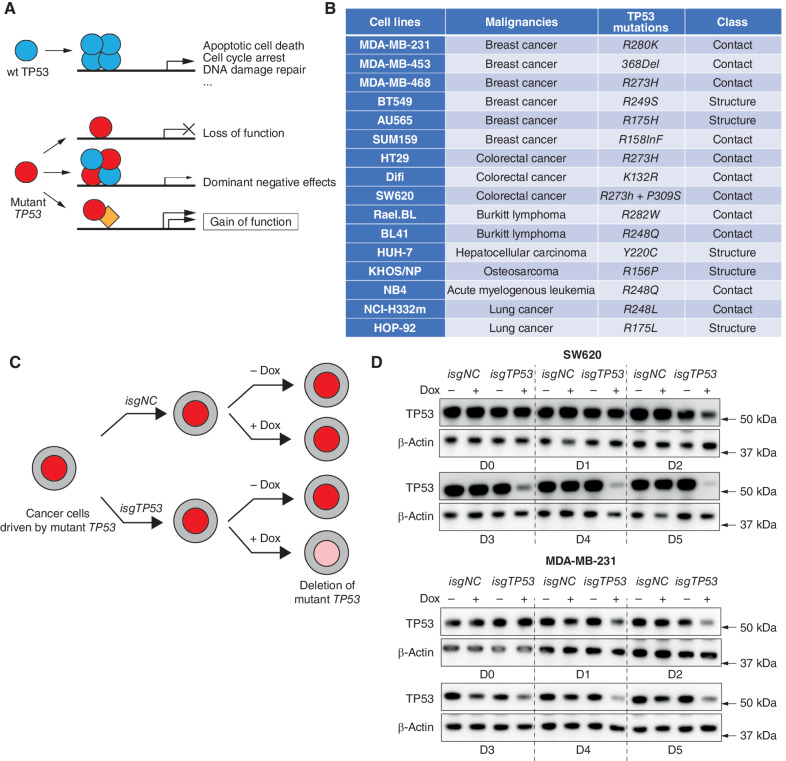
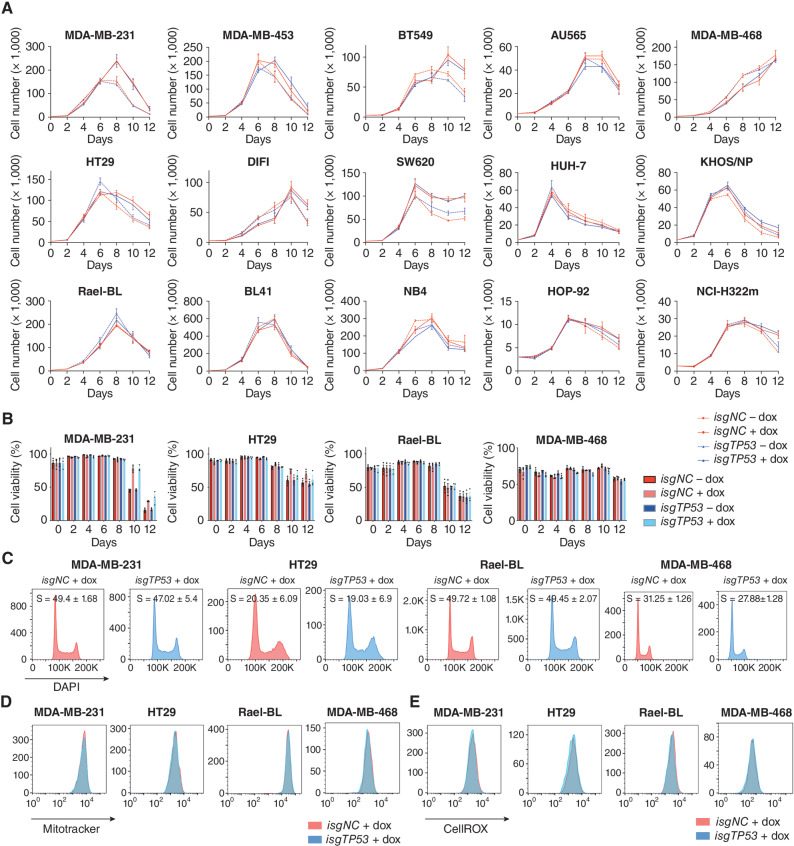
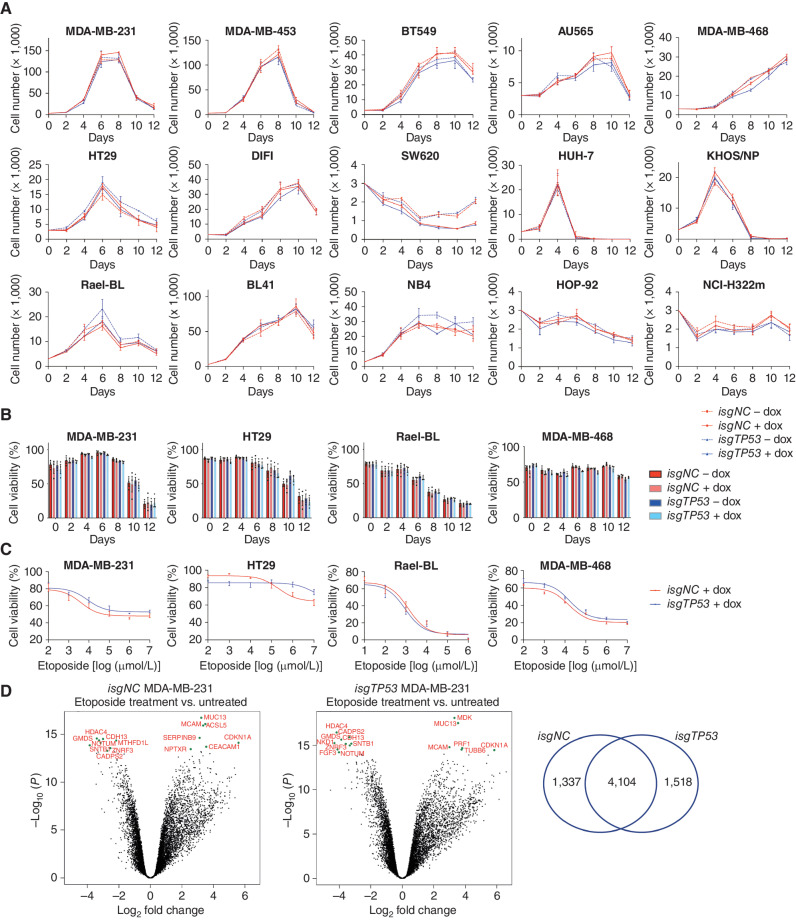
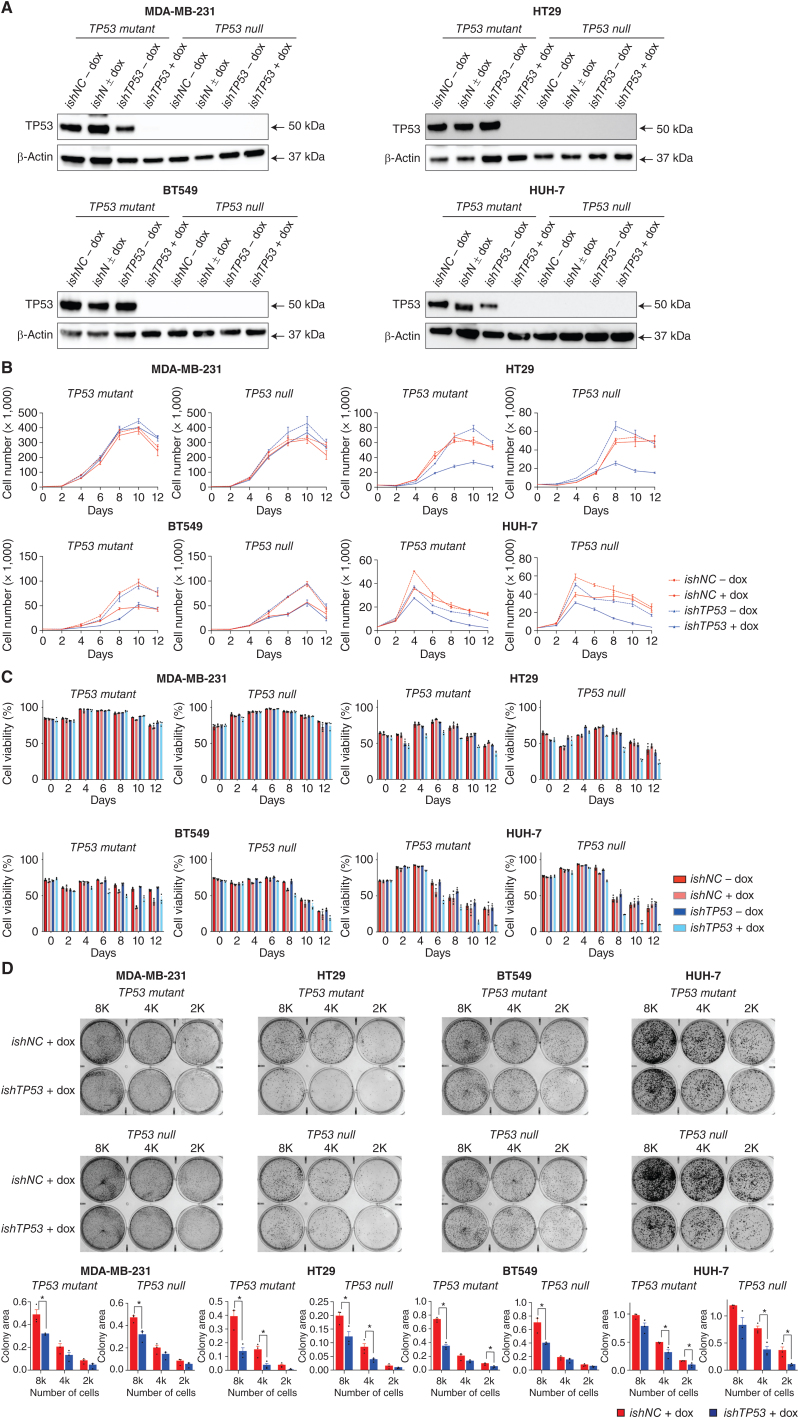
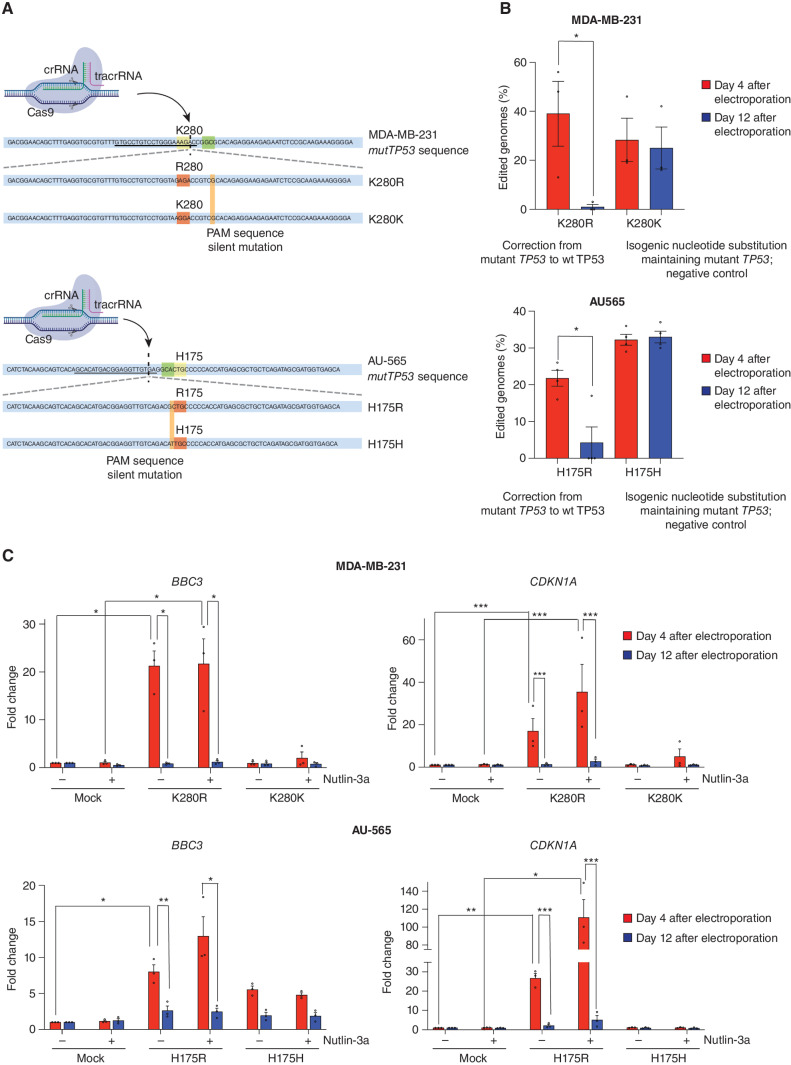
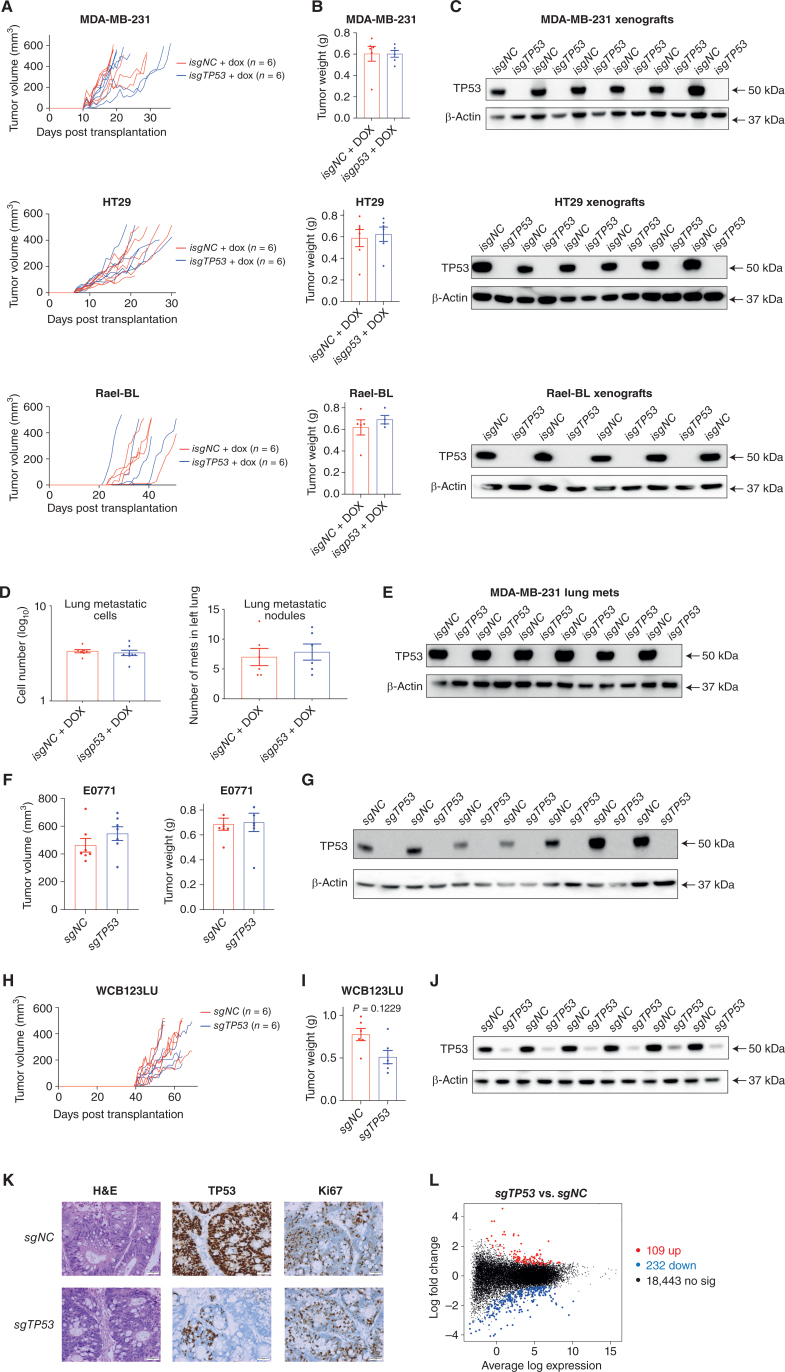
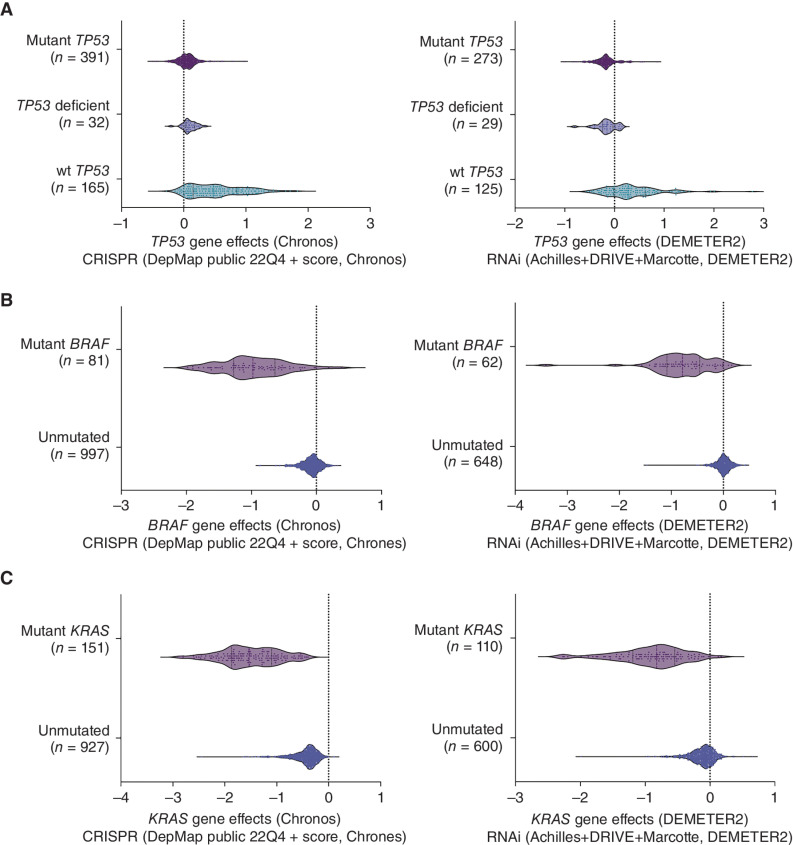
Mutations in the tumor suppressor TP53 cause cancer and impart poor chemotherapeutic responses, reportedly through loss-of-function, dominant-negative effects and gain-of-function (GOF) activities. The relative contributions of these attributes is unknown. We found that removal of 12 different TP53 mutants with reported GOFs by CRISPR/Cas9 did not impact proliferation and response to chemotherapeutics of 15 human cancer cell lines and colon cancer-derived organoids in culture. Moreover, removal of mutant TP53/TRP53 did not impair growth or metastasis of human cancers in immune-deficient mice or growth of murine cancers in immune-competent mice. DepMap mining revealed that removal of 158 different TP53 mutants had no impact on the growth of 391 human cancer cell lines. In contrast, CRISPR-mediated restoration of wild-type TP53 extinguished the growth of human cancer cells in vitro. These findings demonstrate that LOF but not GOF effects of mutant TP53/TRP53 are critical to sustain expansion of many tumor types.
Significance: This study provides evidence that removal of mutant TP53, thereby deleting its reported GOF activities, does not impact the survival, proliferation, metastasis, or chemotherapy responses of cancer cells. Thus, approaches that abrogate expression of mutant TP53 or target its reported GOF activities are unlikely to exert therapeutic impact in cancer. See related commentary by Lane, p. 211 . This article is featured in Selected Articles from This Issue, p. 201.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Aylon Y, Oren M. Living with p53, dying of p53. Cell 2007;130:597–600. - PubMed
-
- Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 2018;24:947–53. - PubMed
-
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007;8:275–83. - PubMed
-
- Vousden KH, Prives C. P53 and prognosis: new insights and further complexity. Cell 2005;120:7–10. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
