

CD19 occupancy with tafasitamab increases therapeutic index of CART19 cell therapy and diminishes severity of CRS
- PMID: 37879074
- PMCID: PMC10808250
- DOI: 10.1182/blood.2022018905
CD19 occupancy with tafasitamab increases therapeutic index of CART19 cell therapy and diminishes severity of CRS
Erratum in
-
Sakemura RL, Manriquez Roman C, Horvei P, et al. CD19 occupancy with tafasitamab increases therapeutic index of CART19 cell therapy and diminishes severity of CRS. Blood. 2024;143(3):258-271.Blood. 2024 Oct 3;144(14):1544. doi: 10.1182/blood.2024026723. Blood. 2024. PMID: 39361306 Free PMC article. No abstract available.
Abstract
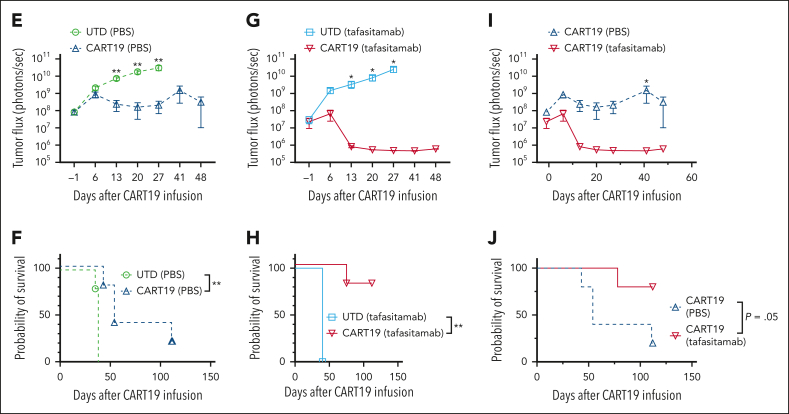
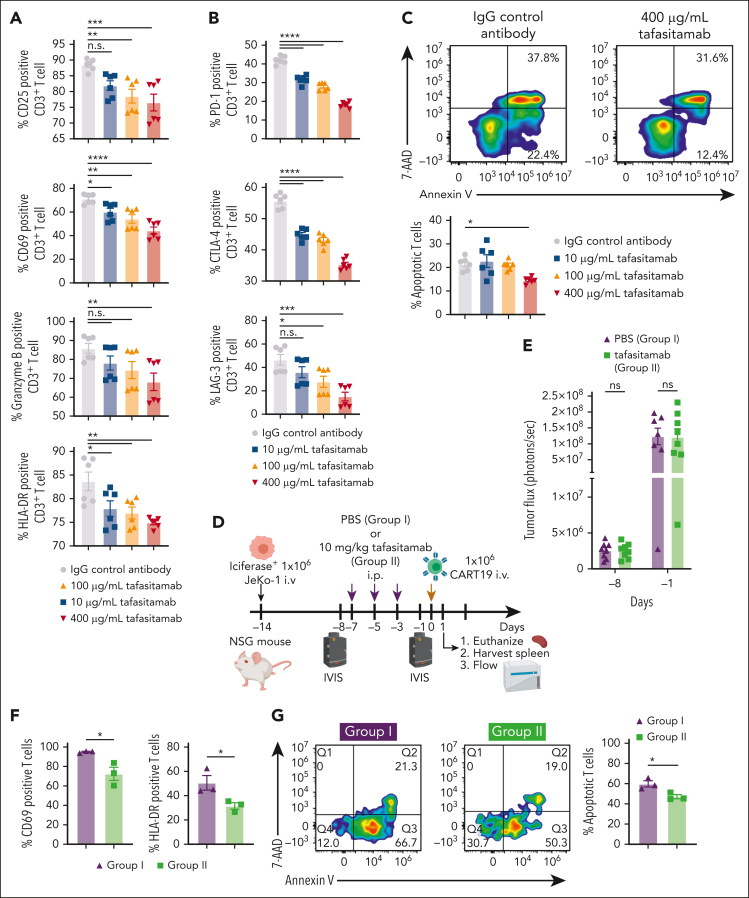
In the development of various strategies of anti-CD19 immunotherapy for the treatment of B-cell malignancies, it remains unclear whether CD19 monoclonal antibody therapy impairs subsequent CD19-targeted chimeric antigen receptor T-cell (CART19) therapy. We evaluated the potential interference between the CD19-targeting monoclonal antibody tafasitamab and CART19 treatment in preclinical models. Concomitant treatment with tafasitamab and CART19 showed major CD19 binding competition, which led to CART19 functional impairment. However, when CD19+ cell lines were pretreated with tafasitamab overnight and the unbound antibody was subsequently removed from the culture, CART19 function was not affected. In preclinical in vivo models, tafasitamab pretreatment demonstrated reduced incidence and severity of cytokine release syndrome and exhibited superior antitumor effects and overall survival compared with CART19 alone. This was associated with transient CD19 occupancy with tafasitamab, which in turn resulted in the inhibition of CART19 overactivation, leading to diminished CAR T apoptosis and pyroptosis of tumor cells.
© 2024 American Society of Hematology. Published by Elsevier Inc. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: S.S.K. is an inventor of patents in the field of chimeric antigen receptor (CAR) immunotherapy that are licensed to Novartis (through an agreement between Mayo Clinic, University of Pennsylvania, and Novartis). R.L.S., and S.S.K. are inventors of patents in the field of CAR immunotherapy that are licensed to Humanigen (through the Mayo Clinic). M.H. and S.S.K. are inventors of patents in the field of CAR immunotherapy that are licensed to Mettaforge (through the Mayo Clinic). S.S.K. receives research funding from Kite, Gilead, Juno, Bristol Myers Squibb (BMS), Novartis, Humanigen, MorphoSys, Tolero, and Lentigen. N.E.K. receives research funding from Acerta Pharma, BMS, Pharmacyclics, MEI Pharma, and Sunesis. N.E.K. has participated in the advisory board meetings of Cytomx Therapy, Janssen, Juno Therapeutics, AstraZeneca, and Oncotracker; and on DSMC for Agios and Cytomx Therapeutics. S.A.P. receives research funding from Pharmacyclics, MorphoSys, Janssen, AstraZeneca, TG Therapeutics, BMS, AbbVie, and Ascentage Pharma. S.A.P. participated in the advisory board meetings of Pharmacyclics, AstraZeneca, Genentech, Gilead, GlaxoSmithKline, Verastem Oncology, and AbbVie (he was not personally compensated for his participation). The remaining authors declare no competing financial interests.
Figures







Comment in
-
CD19 occupancy may drive CARs further.Blood. 2024 Jan 18;143(3):190-192. doi: 10.1182/blood.2023022783. Blood. 2024. PMID: 38236611 No abstract available.
References
-
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767–774. - PubMed
-
- Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. 1989;21(1 pt 1):127–130. - PubMed
-
- Bouchkouj N, Kasamon YL, de Claro RA, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25(6):1702–1708. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
