

Single-cell morphological and topological atlas reveals the ecosystem diversity of human breast cancer
- PMID: 37880211
- PMCID: PMC10600153
- DOI: 10.1038/s41467-023-42504-y
Single-cell morphological and topological atlas reveals the ecosystem diversity of human breast cancer
Abstract
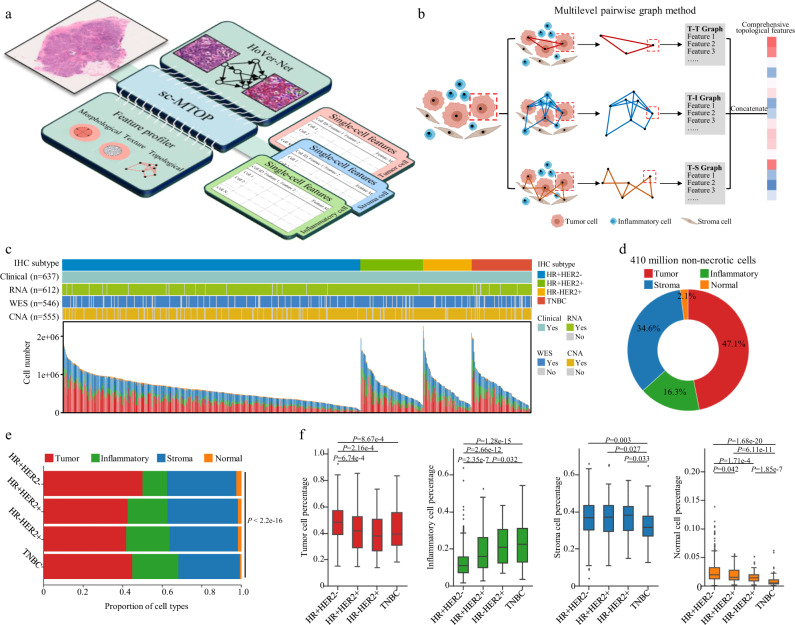
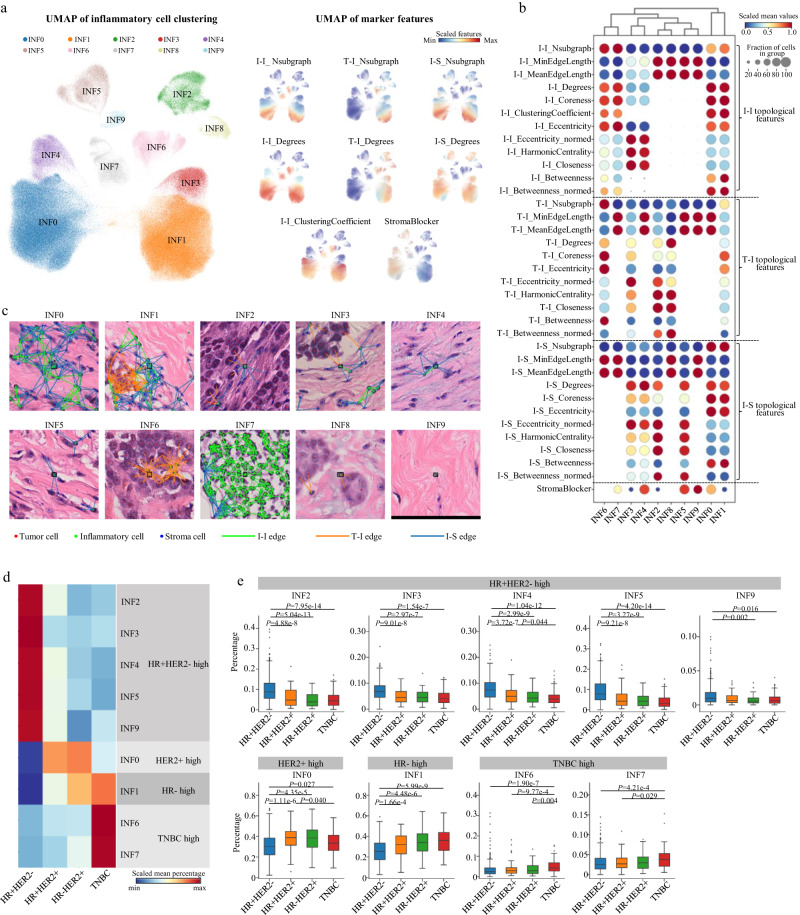
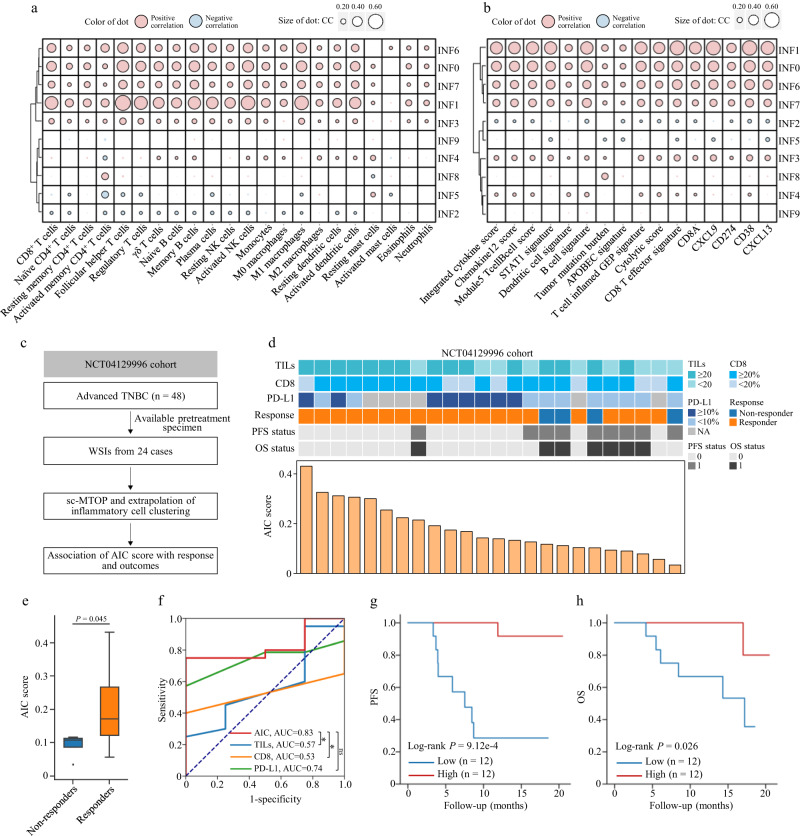
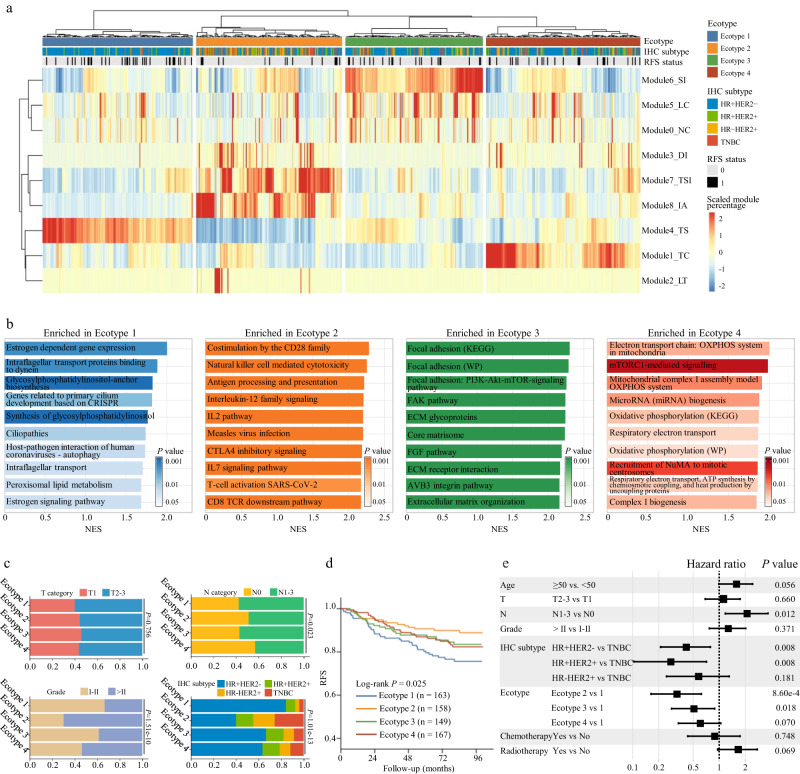
Digital pathology allows computerized analysis of tumor ecosystem using whole slide images (WSIs). Here, we present single-cell morphological and topological profiling (sc-MTOP) to characterize tumor ecosystem by extracting the features of nuclear morphology and intercellular spatial relationship for individual cells. We construct a single-cell atlas comprising 410 million cells from 637 breast cancer WSIs and dissect the phenotypic diversity within tumor, inflammatory and stroma cells respectively. Spatially-resolved analysis identifies recurrent micro-ecological modules representing locoregional multicellular structures and reveals four breast cancer ecotypes correlating with distinct molecular features and patient prognosis. Further analysis with multiomics data uncovers clinically relevant ecosystem features. High abundance of locally-aggregated inflammatory cells indicates immune-activated tumor microenvironment and favorable immunotherapy response in triple-negative breast cancers. Morphological intratumor heterogeneity of tumor nuclei correlates with cell cycle pathway activation and CDK inhibitors responsiveness in hormone receptor-positive cases. sc-MTOP enables using WSIs to characterize tumor ecosystems at the single-cell level.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J. Clin. Oncol. 2010;28:4022–4028. - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer. 2006;6:924–935. - PubMed
-
- Salgado R, Loi S. Tumour infiltrating lymphocytes in breast cancer: increasing clinical relevance. Lancet Oncol. 2018;19:3–5. - PubMed
-
- Schmid P, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018;379:2108–2121. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
