

Diverse electron carriers drive syntrophic interactions in an enriched anaerobic acetate-oxidizing consortium
- PMID: 37880541
- PMCID: PMC10689502
- DOI: 10.1038/s41396-023-01542-6
Diverse electron carriers drive syntrophic interactions in an enriched anaerobic acetate-oxidizing consortium
Abstract
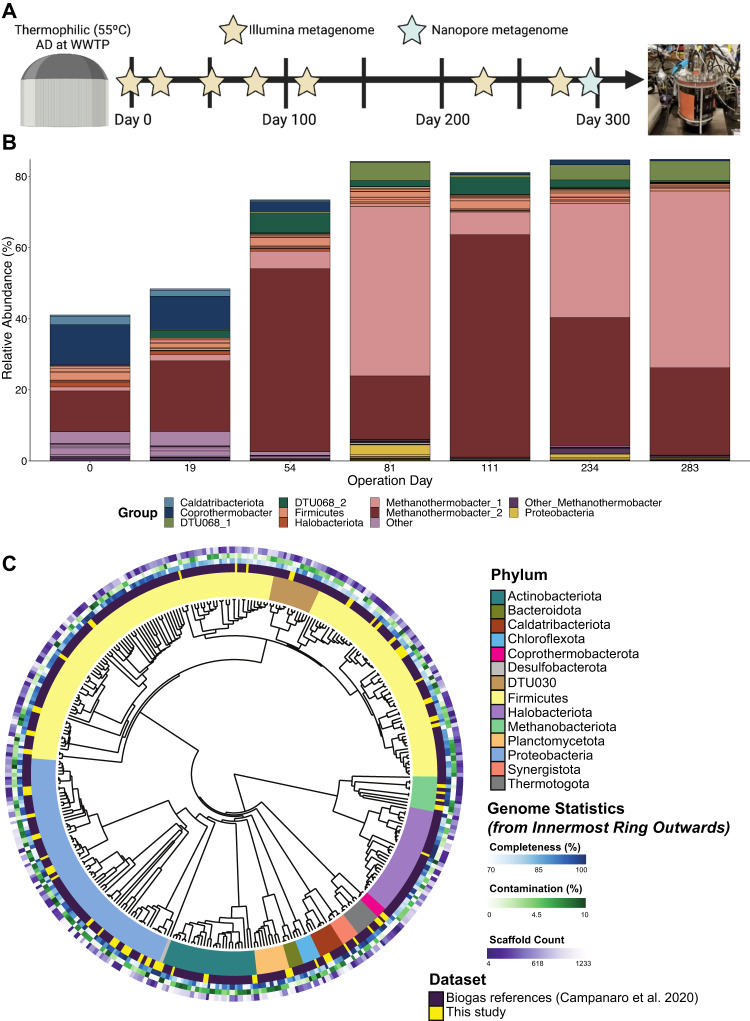
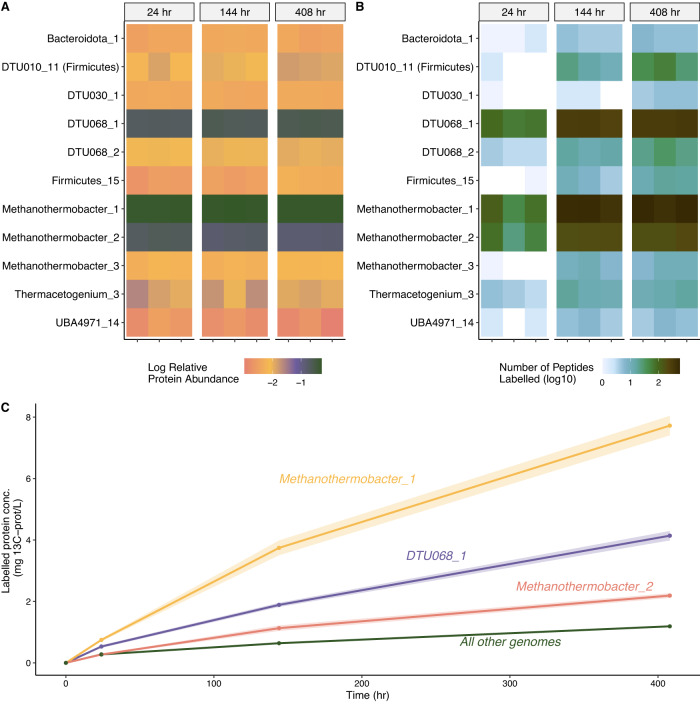
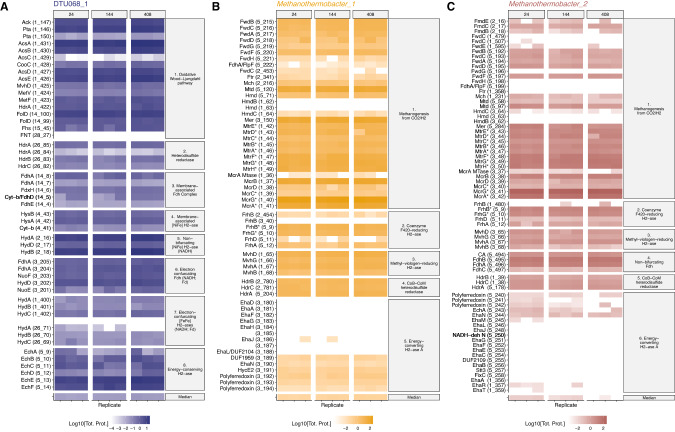
In many anoxic environments, syntrophic acetate oxidation (SAO) is a key pathway mediating the conversion of acetate into methane through obligate cross-feeding interactions between SAO bacteria (SAOB) and methanogenic archaea. The SAO pathway is particularly important in engineered environments such as anaerobic digestion (AD) systems operating at thermophilic temperatures and/or with high ammonia. Despite the widespread importance of SAOB to the stability of the AD process, little is known about their in situ physiologies due to typically low biomass yields and resistance to isolation. Here, we performed a long-term (300-day) continuous enrichment of a thermophilic (55 °C) SAO community from a municipal AD system using acetate as the sole carbon source. Over 80% of the enriched bioreactor metagenome belonged to a three-member consortium, including an acetate-oxidizing bacterium affiliated with DTU068 encoding for carbon dioxide, hydrogen, and formate production, along with two methanogenic archaea affiliated with Methanothermobacter_A. Stable isotope probing was coupled with metaproteogenomics to quantify carbon flux into each community member during acetate conversion and inform metabolic reconstruction and genome-scale modeling. This effort revealed that the two Methanothermobacter_A species differed in their preferred electron donors, with one possessing the ability to grow on formate and the other only consuming hydrogen. A thermodynamic analysis suggested that the presence of the formate-consuming methanogen broadened the environmental conditions where ATP production from SAO was favorable. Collectively, these results highlight how flexibility in electron partitioning during SAO likely governs community structure and fitness through thermodynamic-driven mutualism, shedding valuable insights into the metabolic underpinnings of this key functional group within methanogenic ecosystems.
© 2023. The Author(s).
Conflict of interest statement
With the exception of SJH, the authors declare no competing interests associated with this work. SJH is a co-founder of Koonkie Inc., a bioinformatics consulting company that designs and provides scalable algorithmic and data analytics solutions in the cloud. Koonkie Inc. was not involved in any aspect of this research.
Figures




References
-
- Conrad R. Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments. FEMS Microbiol Ecol. 1999;28:193–202. doi: 10.1111/j.1574-6941.1999.tb00575.x. - DOI
-
- Jetten MSM, Stams AJM, Zehnder AJB. Methanogenesis from acetate: a comparison of the acetate metabolism in Methanothrix soehngenii and Methanosarcina spp. FEMS Microbiol Lett. 1992;88:181–97. doi: 10.1111/j.1574-6968.1992.tb04987.x. - DOI
-
- Zinder SH, Koch M. Non-aceticlastic methanogenesis from acetate: acetate oxidation by a thermophilic syntrophic coculture. Arch Microbiol. 1984;138:263–72. doi: 10.1007/BF00402133. - DOI
