

Extension of the Segatella copri complex to 13 species with distinct large extrachromosomal elements and associations with host conditions
- PMID: 37883976
- PMCID: PMC10635906
- DOI: 10.1016/j.chom.2023.09.013
Extension of the Segatella copri complex to 13 species with distinct large extrachromosomal elements and associations with host conditions
Abstract
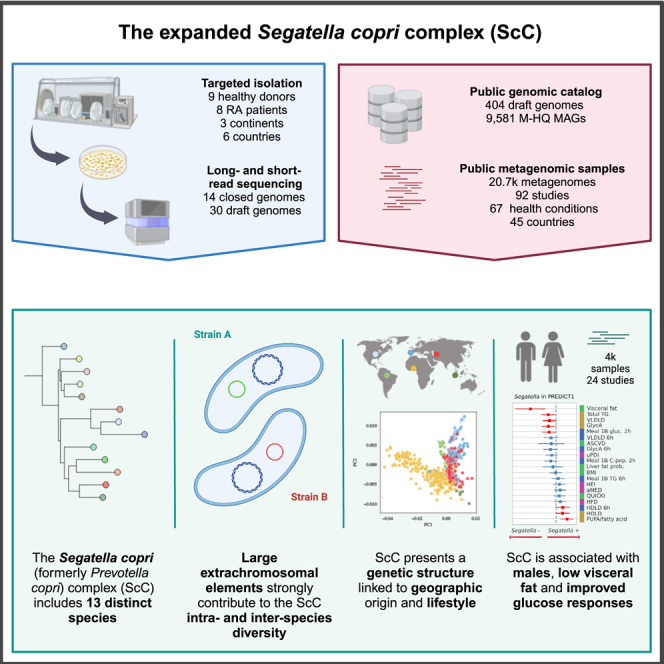
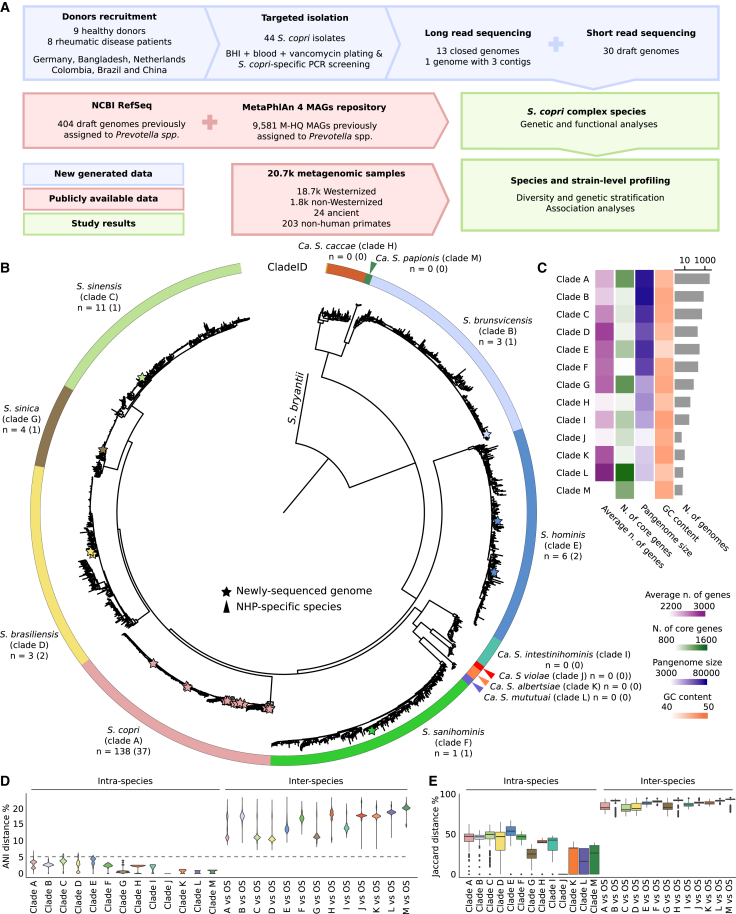
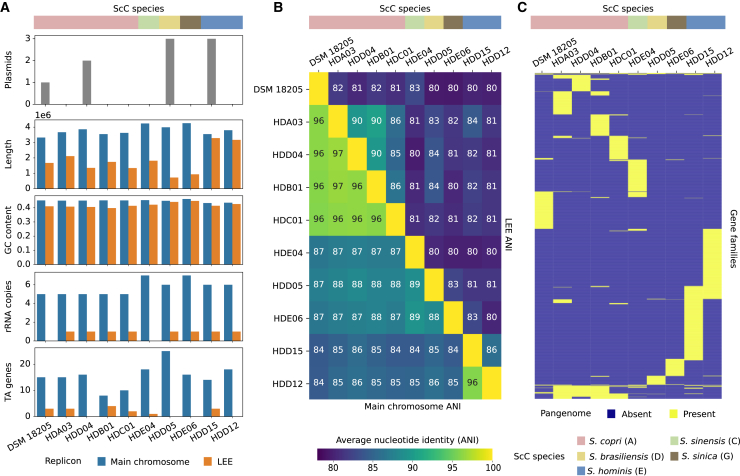
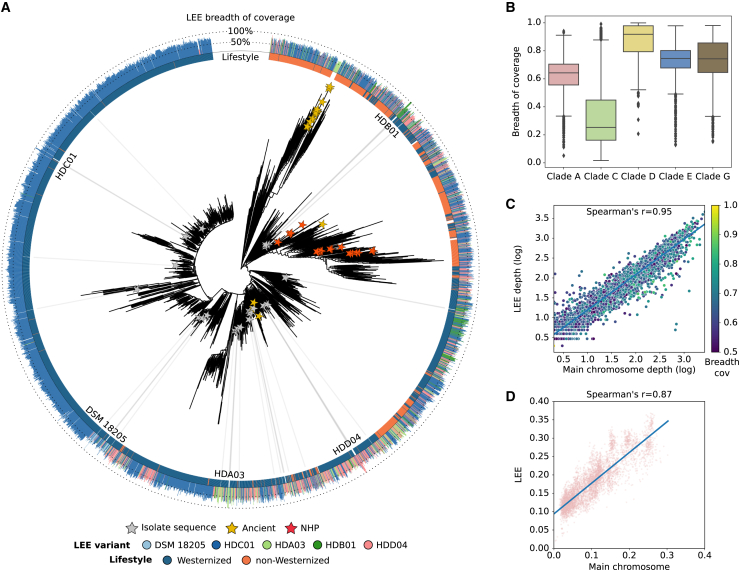
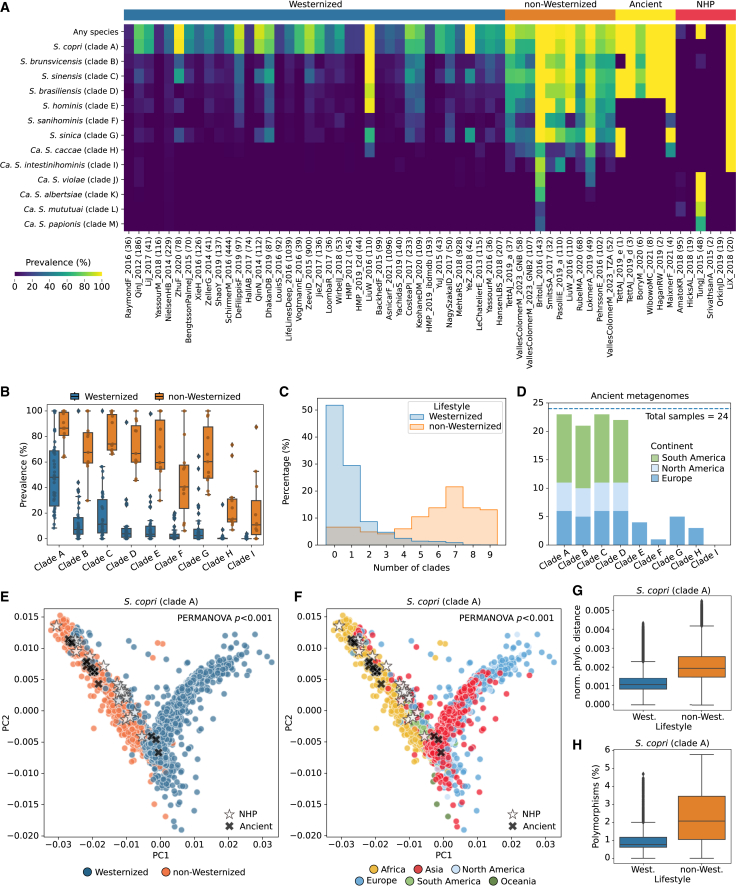
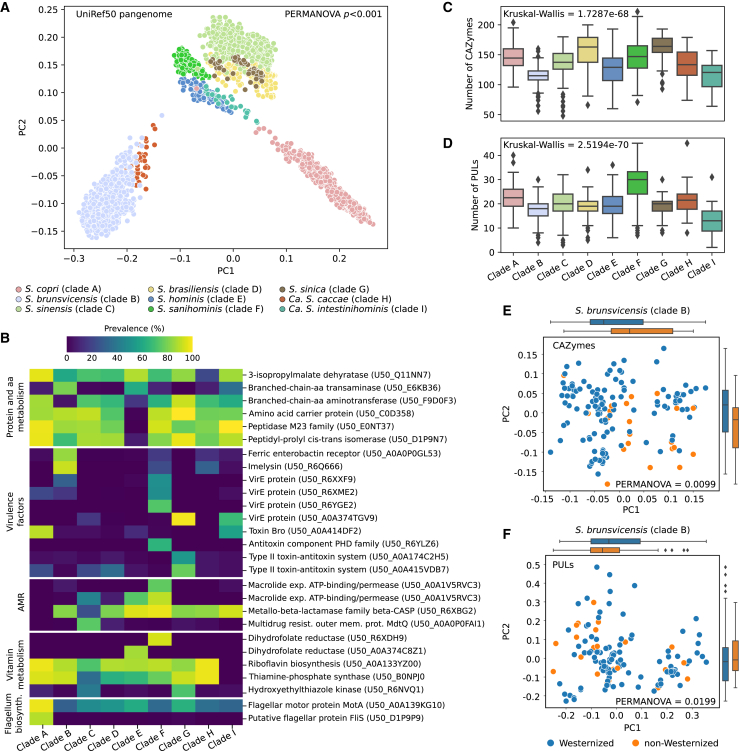
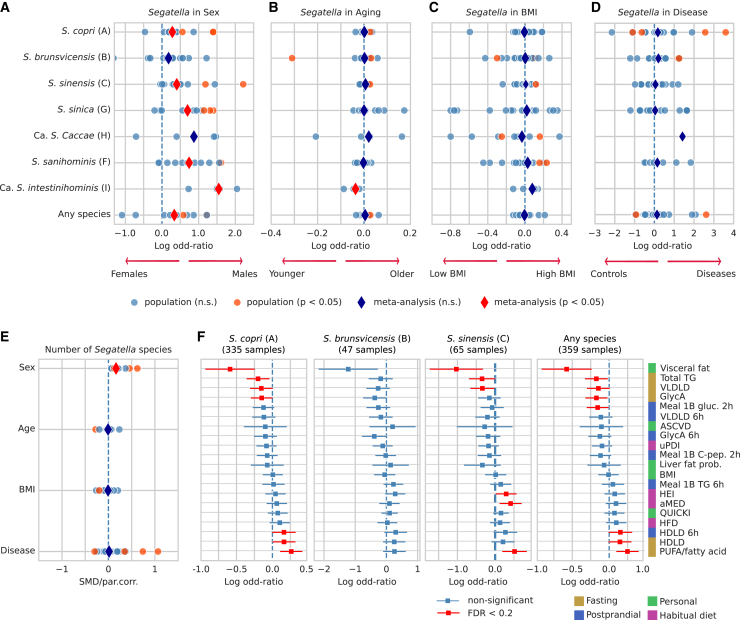
The Segatella copri (formerly Prevotella copri) complex (ScC) comprises taxa that are key members of the human gut microbiome. It was previously described to contain four distinct phylogenetic clades. Combining targeted isolation with large-scale metagenomic analysis, we defined 13 distinct Segatella copri-related species, expanding the ScC complex beyond four clades. Complete genome reconstruction of thirteen strains from seven species unveiled the presence of genetically diverse large circular extrachromosomal elements. These elements are consistently present in most ScC species, contributing to intra- and inter-species diversities. The nine species-level clades present in humans display striking differences in prevalence and intra-species genetic makeup across human populations. Based on a meta-analysis, we found reproducible associations between members of ScC and the male sex and positive correlations with lower visceral fat and favorable markers of cardiometabolic health. Our work uncovers genomic diversity within ScC, facilitating a better characterization of the human microbiome.
Keywords: Prevotella copri; ScC; Segatella copri; bacterial isolation; cardiometabolic health; extrachromosomal element; human microbiome; metagenomics.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests T.D.S. and J.W. are co-founders of ZOE Ltd. (ZOE). S.E.B. and T.D.S. receive payments as consultants to ZOE. R.D. is employed by ZOE. J.W., R.D., S.E.B., and T.D.S. receive options in ZOE.
Figures
References
-
- Blanco-Míguez A., Beghini F., Cumbo F., McIver L.J., Thompson K.N., Zolfo M., Manghi P., Dubois L., Huang K.D., Thomas A.M., et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023;4 doi: 10.1038/s41587-023-01688-w. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
