

An atlas of continuous adaptive evolution in endemic human viruses
- PMID: 37883977
- PMCID: PMC12129310
- DOI: 10.1016/j.chom.2023.09.012
An atlas of continuous adaptive evolution in endemic human viruses
Abstract
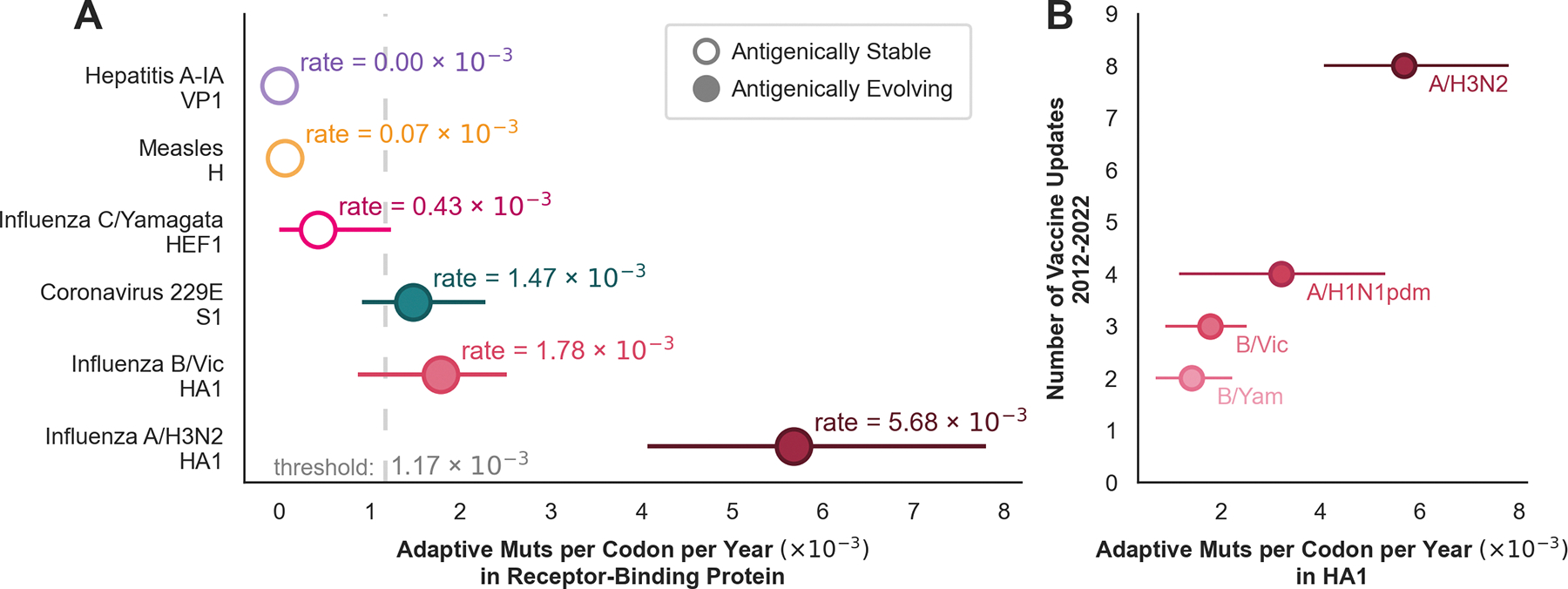
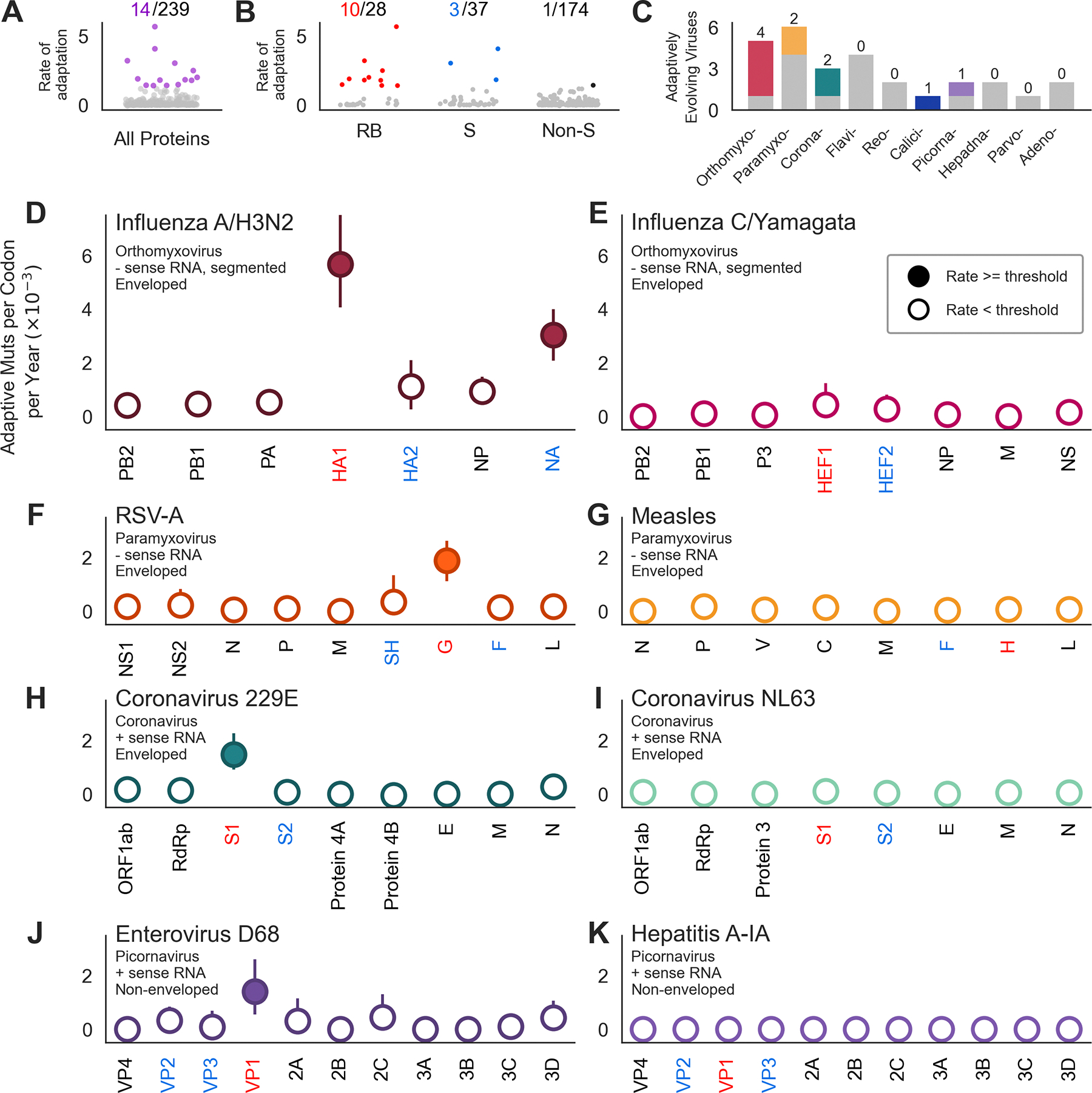
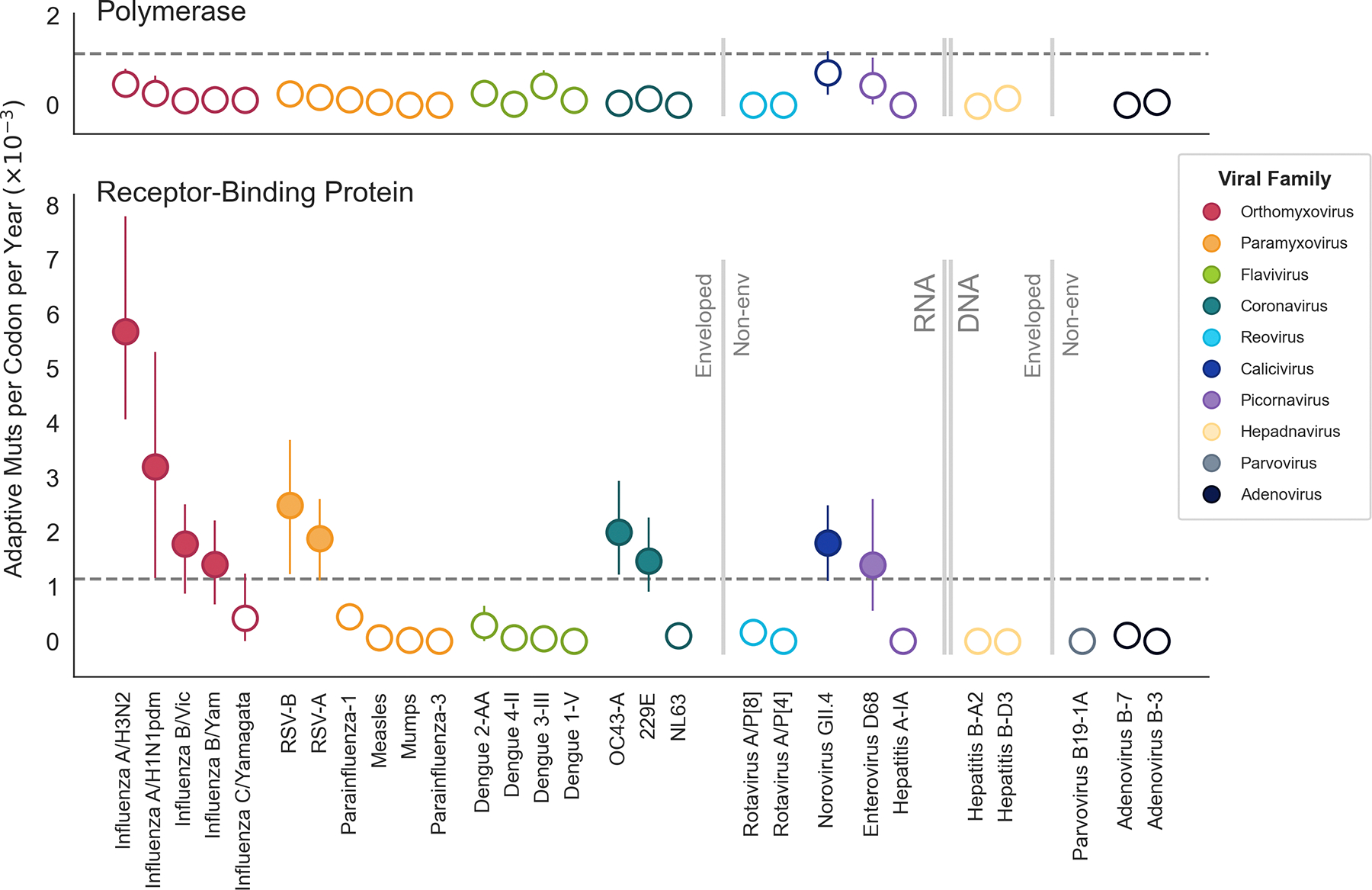
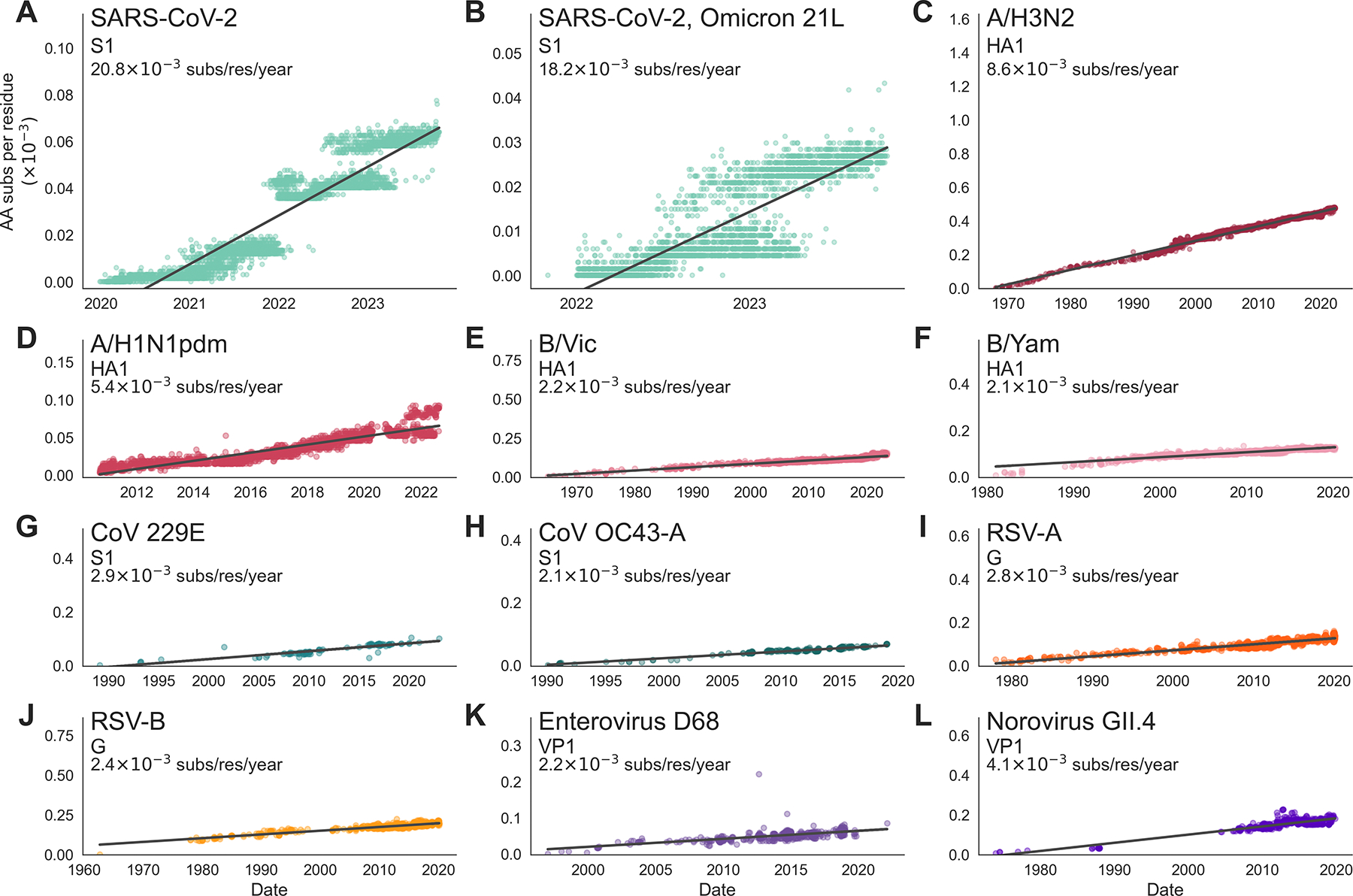
Through antigenic evolution, viruses such as seasonal influenza evade recognition by neutralizing antibodies. This means that a person with antibodies well tuned to an initial infection will not be protected against the same virus years later and that vaccine-mediated protection will decay. To expand our understanding of which endemic human viruses evolve in this fashion, we assess adaptive evolution across the genome of 28 endemic viruses spanning a wide range of viral families and transmission modes. Surface proteins consistently show the highest rates of adaptation, and ten viruses in this panel are estimated to undergo antigenic evolution to selectively fix mutations that enable the escape of prior immunity. Thus, antibody evasion is not an uncommon evolutionary strategy among human viruses, and monitoring this evolution will inform future vaccine efforts. Additionally, by comparing overall amino acid substitution rates, we show that SARS-CoV-2 is accumulating protein-coding changes at substantially faster rates than endemic viruses.
Keywords: adaptive evolution; antigenic evolution; endemic viruses; evolutionary biology; viral evolution.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


Similar articles
-
Immune Escape Adaptive Mutations in Hemagglutinin Are Responsible for the Antigenic Drift of Eurasian Avian-Like H1N1 Swine Influenza Viruses.J Virol. 2022 Aug 24;96(16):e0097122. doi: 10.1128/jvi.00971-22. Epub 2022 Aug 2. J Virol. 2022. PMID: 35916512 Free PMC article.
-
Cross-Protection by Inactivated H5 Prepandemic Vaccine Seed Strains against Diverse Goose/Guangdong Lineage H5N1 Highly Pathogenic Avian Influenza Viruses.J Virol. 2020 Nov 23;94(24):e00720-20. doi: 10.1128/JVI.00720-20. Print 2020 Nov 23. J Virol. 2020. PMID: 32999029 Free PMC article.
-
Mutations in the Hemagglutinin Stalk Domain Do Not Permit Escape from a Protective, Stalk-Based Vaccine-Induced Immune Response in the Mouse Model.mBio. 2021 Feb 16;12(1):e03617-20. doi: 10.1128/mBio.03617-20. mBio. 2021. PMID: 33593972 Free PMC article.
-
Broadly neutralizing antibodies to combat influenza virus infection.Antiviral Res. 2024 Jan;221:105785. doi: 10.1016/j.antiviral.2023.105785. Epub 2023 Dec 23. Antiviral Res. 2024. PMID: 38145757 Review.
-
The antigenic architecture of the hemagglutinin of influenza H5N1 viruses.Mol Immunol. 2013 Dec;56(4):705-19. doi: 10.1016/j.molimm.2013.07.010. Epub 2013 Aug 7. Mol Immunol. 2013. PMID: 23933511 Review.
Cited by
-
RSV F evolution escapes some monoclonal antibodies but does not strongly erode neutralization by human polyclonal sera.J Virol. 2025 Jul 22;99(7):e0053125. doi: 10.1128/jvi.00531-25. Epub 2025 Jul 3. J Virol. 2025. PMID: 40607811 Free PMC article.
-
Fitness models provide accurate short-term forecasts of SARS-CoV-2 variant frequency.PLoS Comput Biol. 2024 Sep 6;20(9):e1012443. doi: 10.1371/journal.pcbi.1012443. eCollection 2024 Sep. PLoS Comput Biol. 2024. PMID: 39241101 Free PMC article.
-
Disruption of seasonal influenza circulation and evolution during the 2009 H1N1 and COVID-19 pandemics in Southeastern Asia.Nat Commun. 2025 Jan 8;16(1):475. doi: 10.1038/s41467-025-55840-y. Nat Commun. 2025. PMID: 39774646 Free PMC article.
-
Frequency dynamics predict viral fitness, antigenic relationships and epidemic growth.medRxiv [Preprint]. 2025 Jan 23:2024.12.02.24318334. doi: 10.1101/2024.12.02.24318334. medRxiv. 2025. PMID: 39677467 Free PMC article. Preprint.
-
H3 hemagglutinin proteins optimized for 2018 to 2022 elicit neutralizing antibodies across panels of modern influenza A(H3N2) viruses.J Immunol. 2025 Jul 1;214(7):1698-1713. doi: 10.1093/jimmun/vkaf092. J Immunol. 2025. PMID: 40400055
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
