

IL-20 controls resolution of experimental colitis by regulating epithelial IFN/STAT2 signalling
- PMID: 37884352
- PMCID: PMC10850655
- DOI: 10.1136/gutjnl-2023-329628
IL-20 controls resolution of experimental colitis by regulating epithelial IFN/STAT2 signalling
Abstract
Objective: We sought to investigate the role of interleukin (IL)-20 in IBD and experimental colitis.
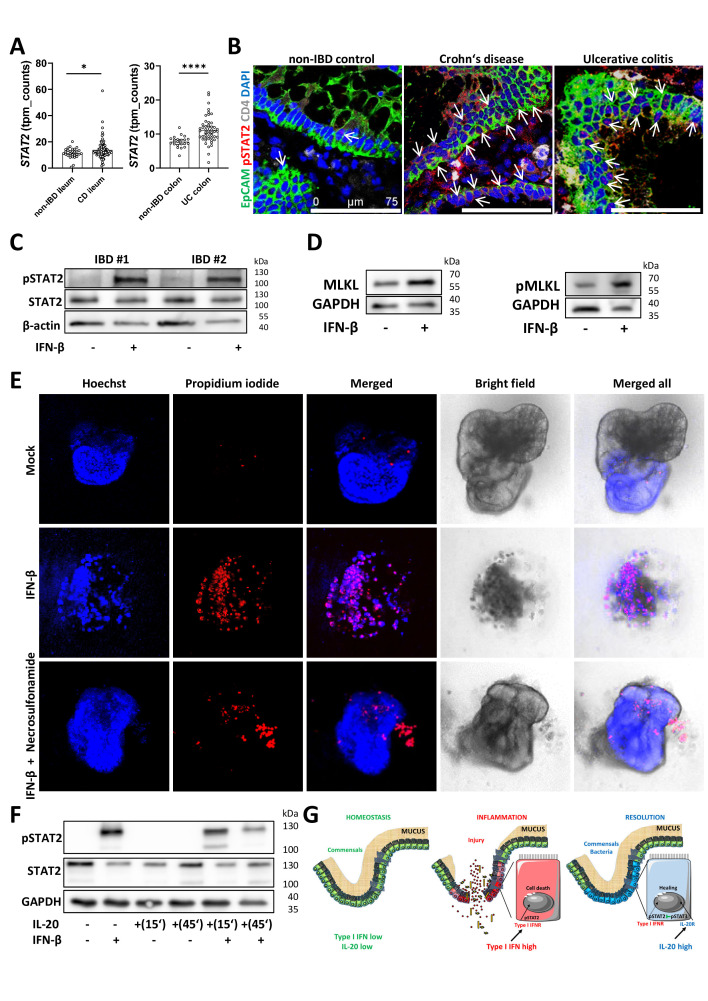
Design: Experimental colitis was induced in mice deficient in components of the IL-20 and signal transducer and activator of transcription (STAT)2 signalling pathways. In vivo imaging, high-resolution mini-endoscopy and histology were used to assess intestinal inflammation. We further used RNA-sequencing (RNA-Seq), RNAScope and Gene Ontology analysis, western blot analysis and co-immunoprecipitation, confocal microscopy and intestinal epithelial cell (IEC)-derived three-dimensional organoids to investigate the underlying molecular mechanisms. Results were validated using samples from patients with IBD and non-IBD control subjects by a combination of RNA-Seq, organoids and immunostainings.
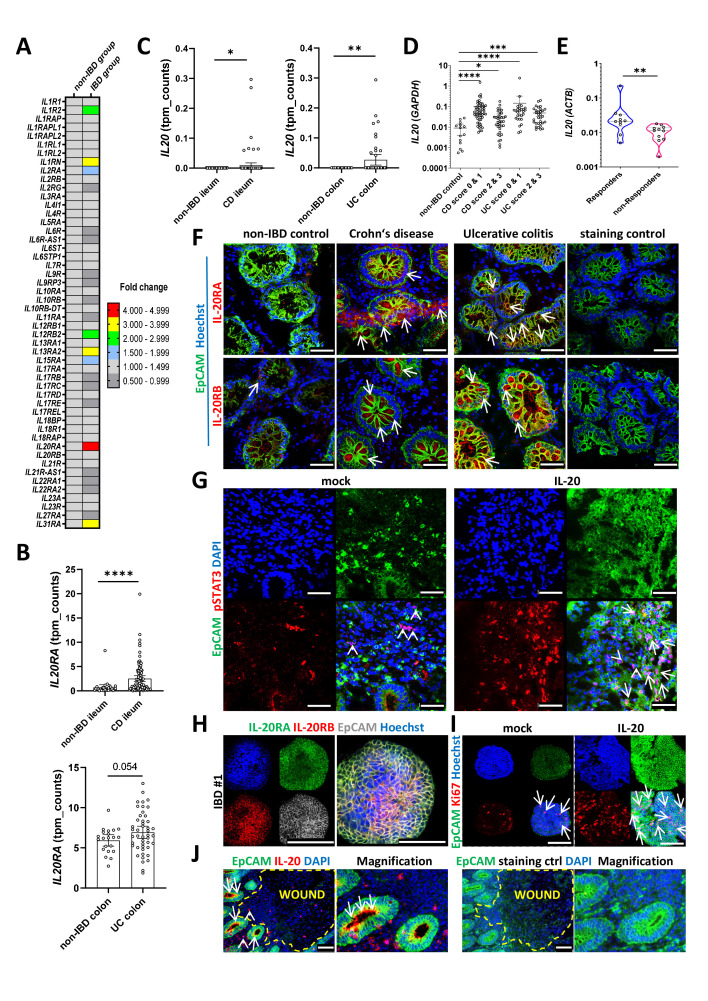
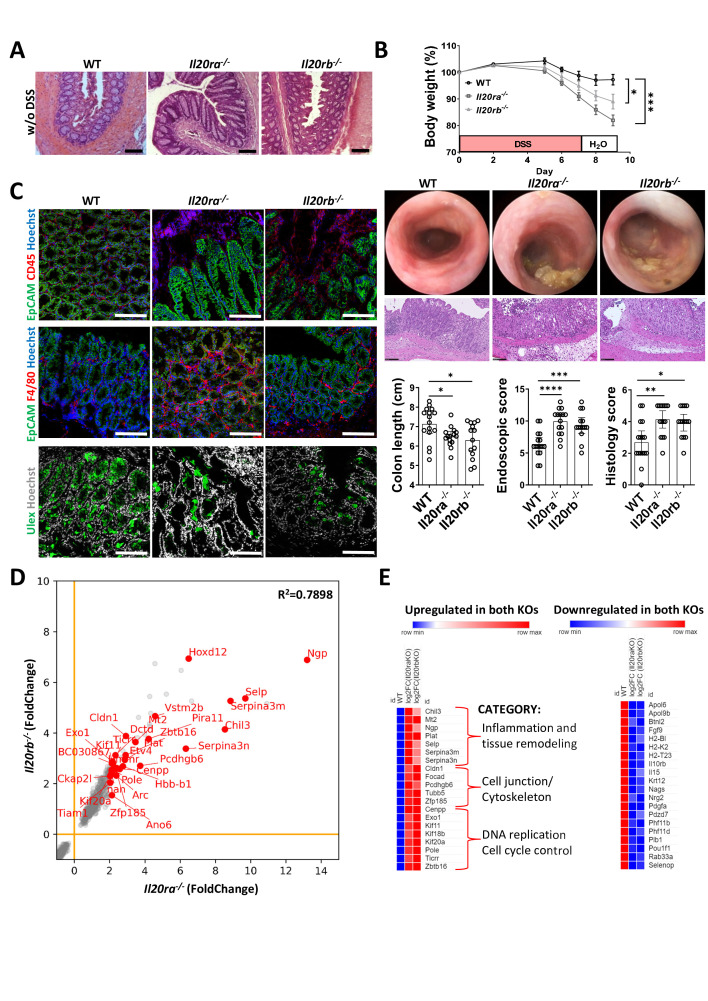
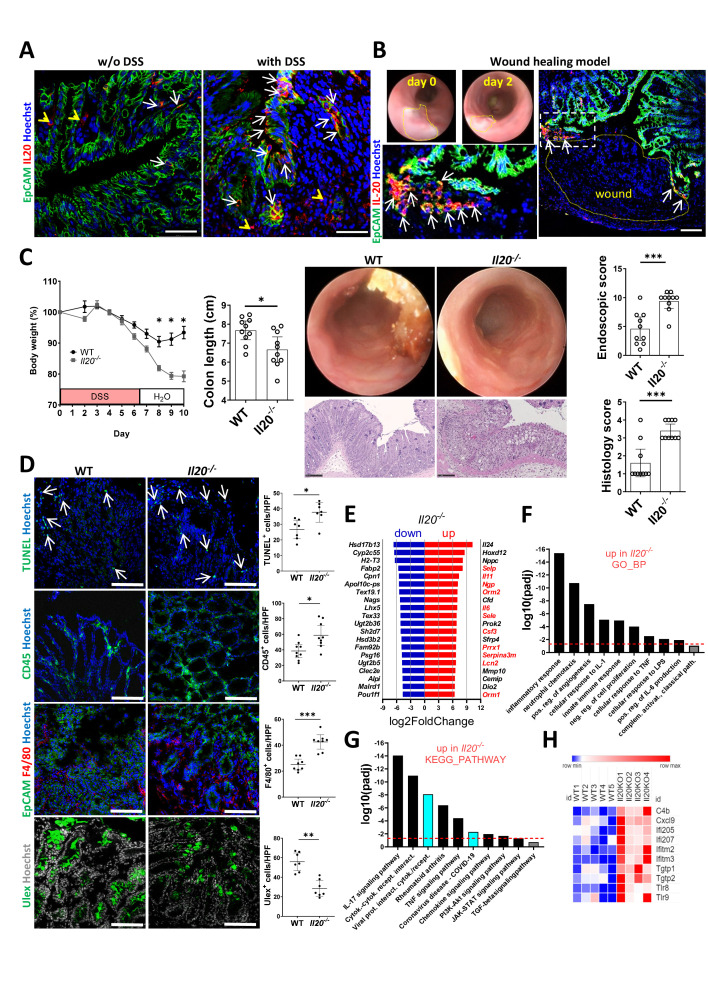
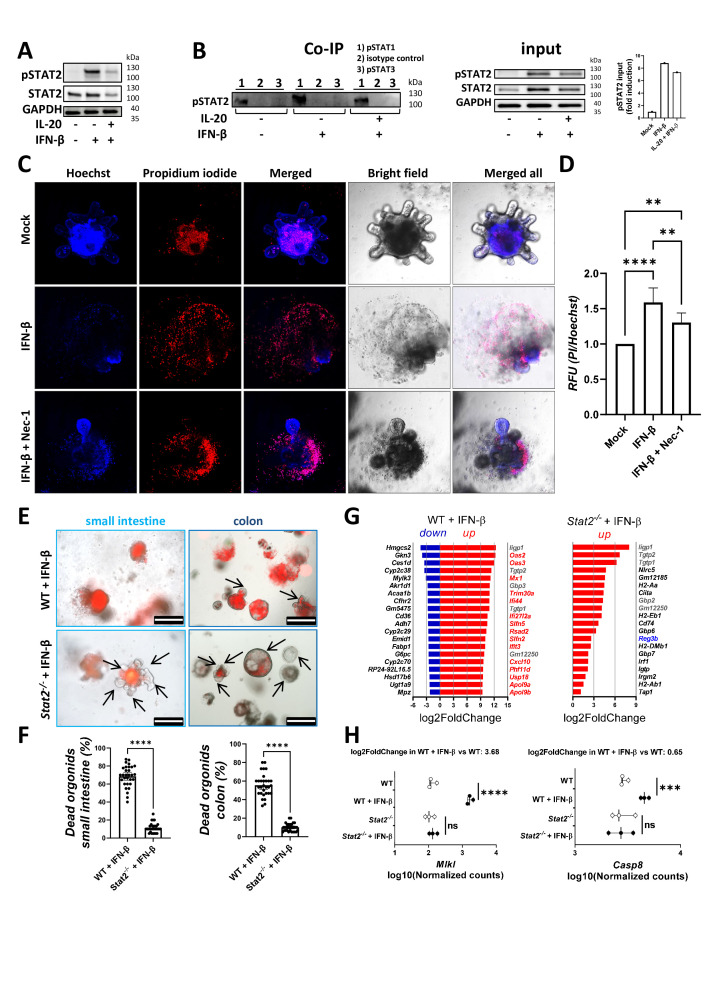
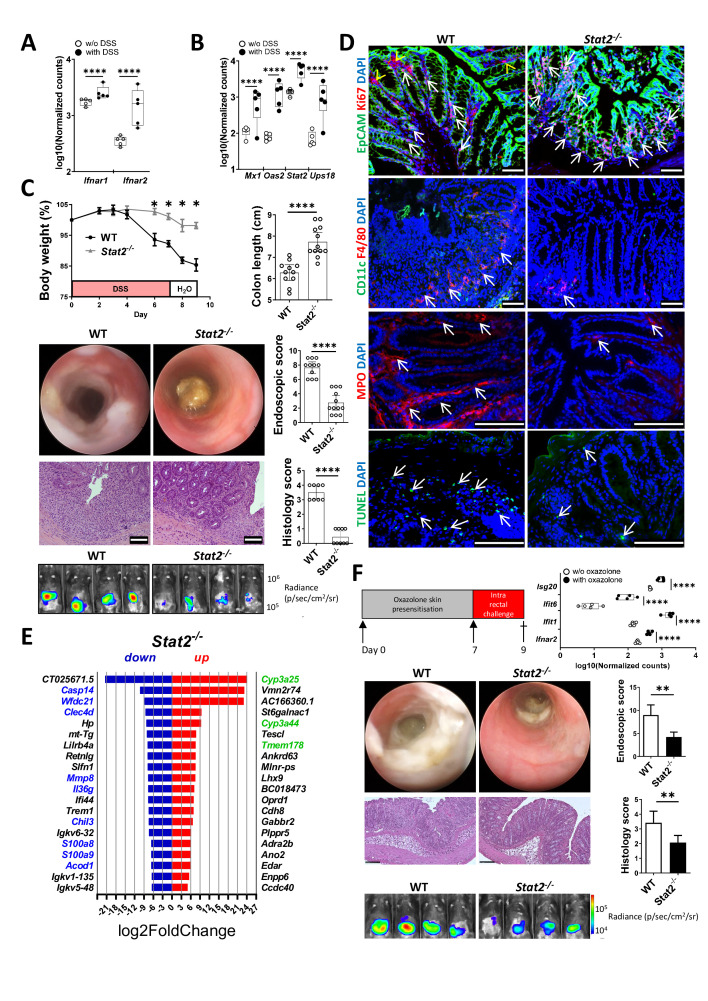
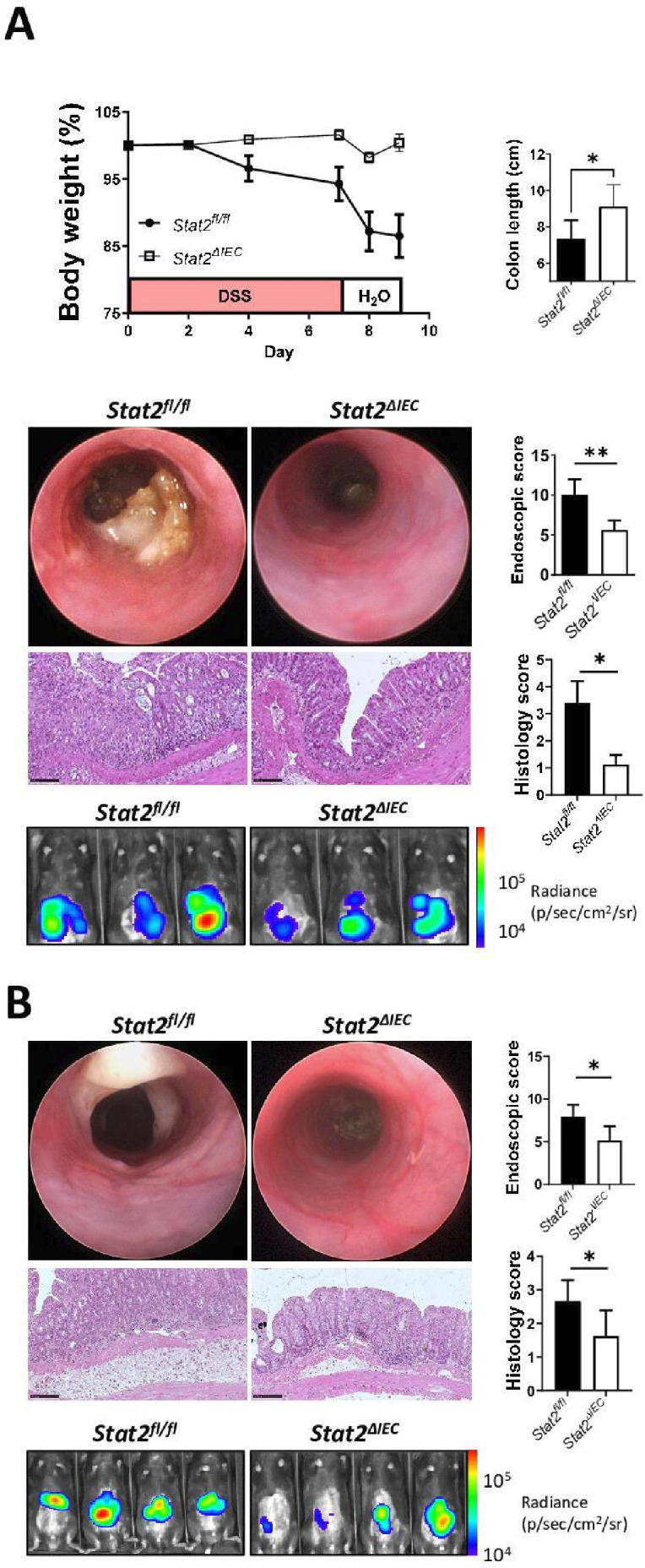
Results: In IBD, IL20 levels were induced during remission and were significantly higher in antitumour necrosis factor responders versus non-responders. IL-20RA and IL-20RB were present on IECs from patients with IBD and IL-20-induced STAT3 and suppressed interferon (IFN)-STAT2 signalling in these cells. In IBD, experimental dextran sulfate sodium (DSS)-induced colitis and mucosal healing, IECs were the main producers of IL-20. Compared with wildtype controls, Il20-/-, Il20ra-/- and Il20rb-/- mice were more susceptible to experimental DSS-induced colitis. IL-20 deficiency was associated with increased IFN/STAT2 activity in mice and IFN/STAT2-induced necroptotic cell death in IEC-derived organoids could be markedly blocked by IL-20. Moreover, newly generated Stat2ΔIEC mice, lacking STAT2 in IECs, were less susceptible to experimental colitis compared with wildtype controls and the administration of IL-20 suppressed colitis activity in wildtype animals.
Conclusion: IL-20 controls colitis and mucosal healing by interfering with the IFN/STAT2 death signalling pathway in IECs. These results indicate new directions for suppressing gut inflammation by modulating IL-20-controlled STAT2 signals.
Keywords: experimental colitis; inflammatory bowel disease; interferon; interleukins; signal transduction.
© Author(s) (or their employer(s)) 2024. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: MFN has served as an advisor for Pentax, Giuliani, PPM, BMS, Janssen, MSD, Takeda and Boehringer. MFN has served as an Associate Editor of the journal/Editorial Board Member. BS has served as consultant for AbbVie, Arena, BMS, Boehringer, Celgene, Falk, Galapagos, Janssen, Lilly, Pfizer, Prometheus and Takeda and received speaker’s fees from AbbVie, CED Service, Falk, Ferring, Janssen, Novartis, Pfizer, Takeda (served as representative of the Charité). All other authors have nothing to disclose.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous