

NCT/DKFZ MASTER handbook of interpreting whole-genome, transcriptome, and methylome data for precision oncology
- PMID: 37884744
- PMCID: PMC10603123
- DOI: 10.1038/s41698-023-00458-w
NCT/DKFZ MASTER handbook of interpreting whole-genome, transcriptome, and methylome data for precision oncology
Abstract
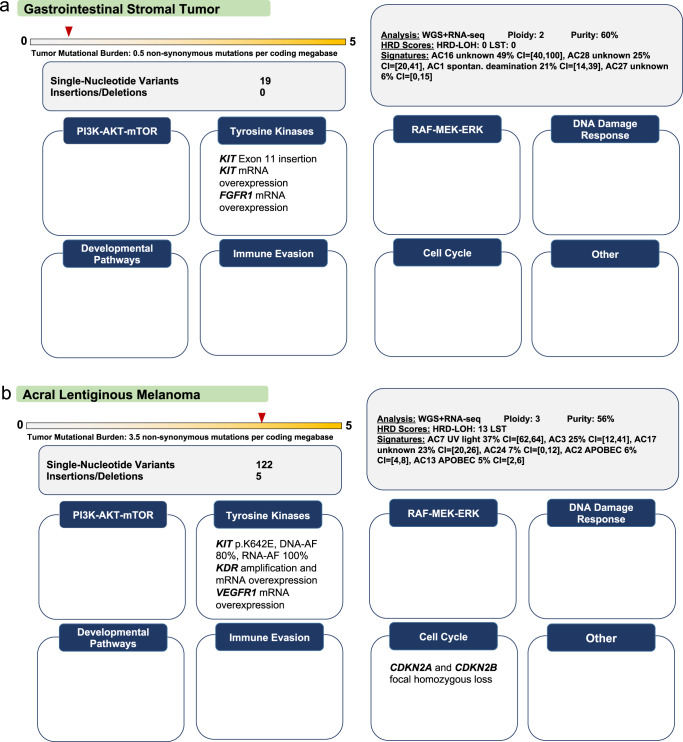
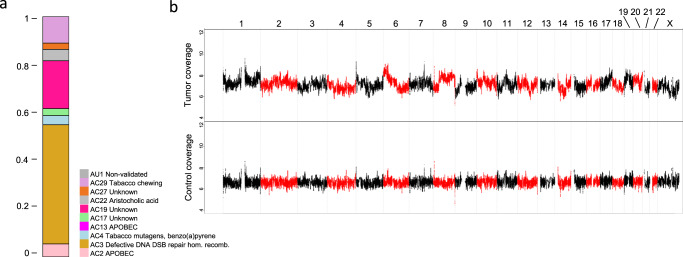
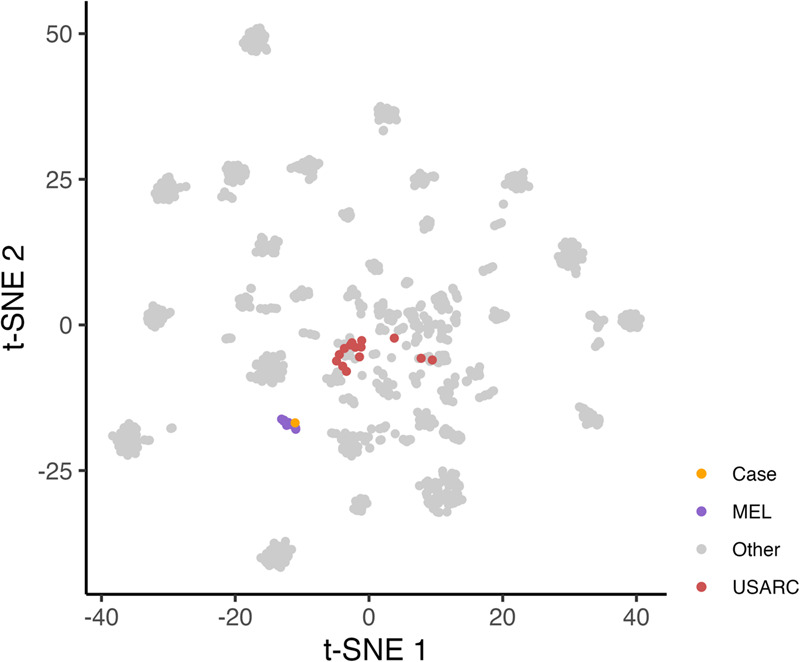
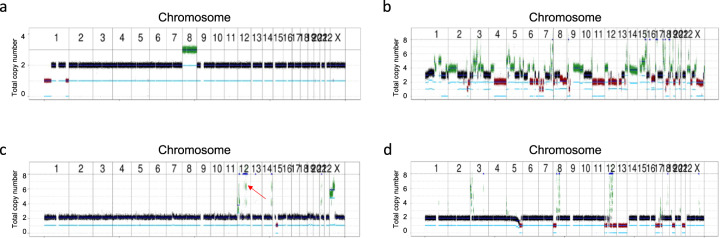
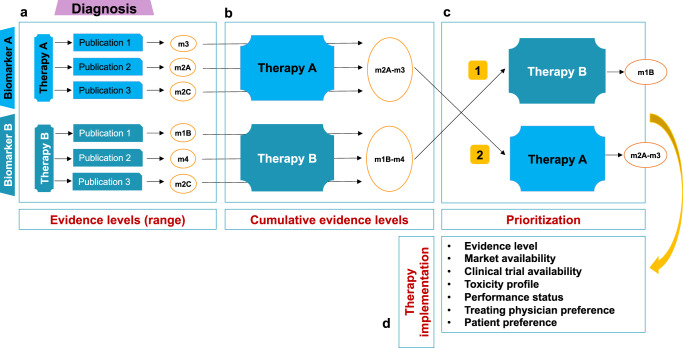
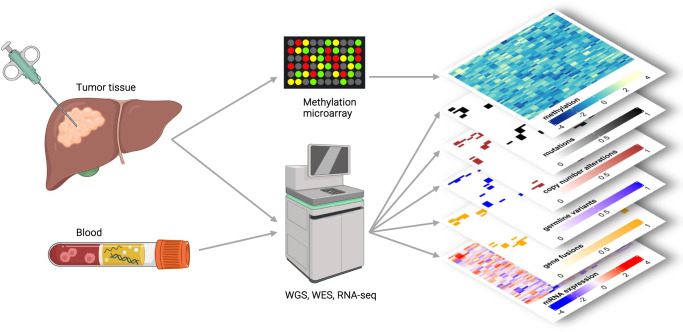
Analysis of selected cancer genes has become an important tool in precision oncology but cannot fully capture the molecular features and, most importantly, vulnerabilities of individual tumors. Observational and interventional studies have shown that decision-making based on comprehensive molecular characterization adds significant clinical value. However, the complexity and heterogeneity of the resulting data are major challenges for disciplines involved in interpretation and recommendations for individualized care, and limited information exists on how to approach multilayered tumor profiles in clinical routine. We report our experience with the practical use of data from whole-genome or exome and RNA sequencing and DNA methylation profiling within the MASTER (Molecularly Aided Stratification for Tumor Eradication Research) program of the National Center for Tumor Diseases (NCT) Heidelberg and Dresden and the German Cancer Research Center (DKFZ). We cover all relevant steps of an end-to-end precision oncology workflow, from sample collection, molecular analysis, and variant prioritization to assigning treatment recommendations and discussion in the molecular tumor board. To provide insight into our approach to multidimensional tumor profiles and guidance on interpreting their biological impact and diagnostic and therapeutic implications, we present case studies from the NCT/DKFZ molecular tumor board that illustrate our daily practice. This manual is intended to be useful for physicians, biologists, and bioinformaticians involved in the clinical interpretation of genome-wide molecular information.
© 2023. The Author(s).
Conflict of interest statement
C.E.H.: Consulting or advisory board membership: Boehringer Ingelheim; honoraria: Novartis, Roche; research funding: Boehringer Ingelheim. S.F.: Consulting or advisory board membership: Bayer, Illumina, Roche; honoraria: Amgen, Eli Lilly, PharmaMar, Roche; research funding: AstraZeneca, Pfizer, PharmaMar, Roche; travel or accommodation expenses: Amgen, Eli Lilly, Illumina, PharmaMar, Roche.
Figures

References
LinkOut - more resources
Full Text Sources
