

Mitochondrial modulation with leriglitazone as a potential treatment for Rett syndrome
- PMID: 37884937
- PMCID: PMC10601217
- DOI: 10.1186/s12967-023-04622-5
Mitochondrial modulation with leriglitazone as a potential treatment for Rett syndrome
Abstract
Background: Rett syndrome is a neuropediatric disease occurring due to mutations in MECP2 and characterized by a regression in the neuronal development following a normal postnatal growth, which results in the loss of acquired capabilities such as speech or purposeful usage of hands. While altered neurotransmission and brain development are the center of its pathophysiology, alterations in mitochondrial performance have been previously outlined, shaping it as an attractive target for the disease treatment.
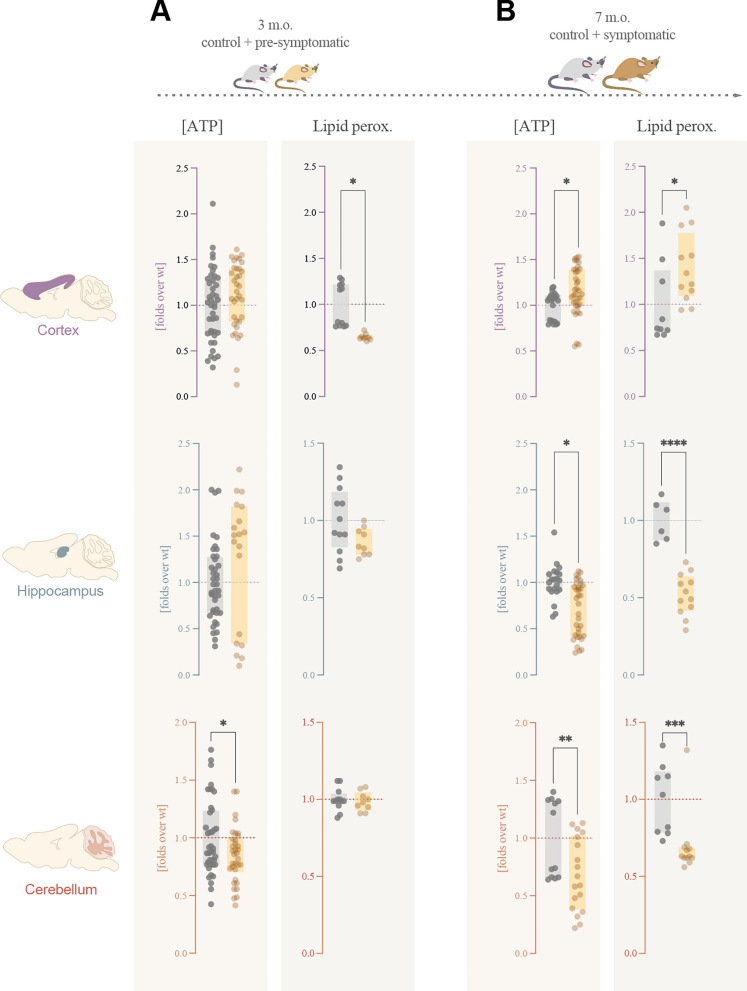
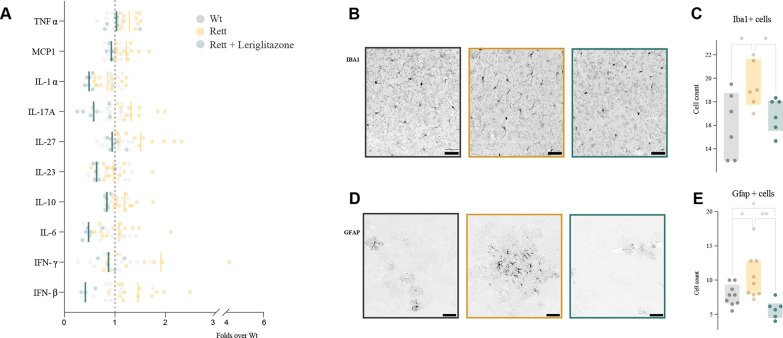
Methods: We have thoroughly described mitochondrial performance in two Rett models, patients' primary fibroblasts and female Mecp2tm1.1Bird-/+ mice brain, discriminating between different brain areas. The characterization was made according to their bioenergetics function, oxidative stress, network dynamics or ultrastructure. Building on that, we have studied the effect of leriglitazone, a PPARγ agonist, in the modulation of mitochondrial performance. For that, we treated Rett female mice with 75 mg/kg/day leriglitazone from weaning until sacrifice at 7 months, studying both the mitochondrial performance changes and their consequences on the mice phenotype. Finally, we studied its effect on neuroinflammation based on the presence of reactive glia by immunohistochemistry and through a cytokine panel.
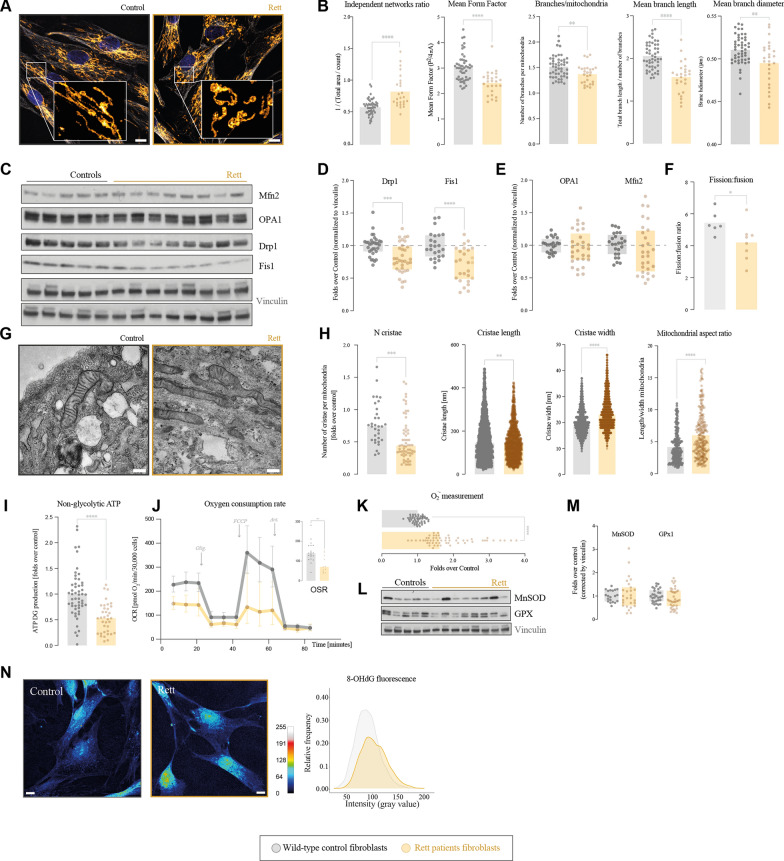
Results: We have described mitochondrial alterations in Rett fibroblasts regarding both shape and bioenergetic functions, as they displayed less interconnected and shorter mitochondria and reduced ATP production along with increased oxidative stress. The bioenergetic alterations were recalled in Rett mice models, being especially significant in cerebellum, already detectable in pre-symptomatic stages. Treatment with leriglitazone recovered the bioenergetic alterations both in Rett fibroblasts and female mice and exerted an anti-inflammatory effect in the latest, resulting in the amelioration of the mice phenotype both in general condition and exploratory activity.
Conclusions: Our studies confirm the mitochondrial dysfunction in Rett syndrome, setting the differences through brain areas and disease stages. Its modulation through leriglitazone is a potential treatment for this disorder, along with other diseases with mitochondrial involvement. This work constitutes the preclinical necessary evidence to lead to a clinical trial.
Keywords: Bioenergetics; Leriglitazone; Metabolic modulation; Mitochondria; Neuroinflammation; Rett syndrome.
© 2023. The Author(s).
Conflict of interest statement
Pilar Pizcueta, Cristina Vergara, Laura Rodríguez-Pascau and Marc Martinell are staff from the pharmaceutic company Minoryx Therapeutics. Marc Martinell and Pilar Pizcueta report a patent application (10198126) issued on January 15th 2019 for the use of leriglitazone described in this paper. The patent will be owned by those authors. Marc Martinell is the cofounder of Minoryx Threapeutics, focused on the development of therapies for rare neurodegenerative diseases. The rest of the authors declare no competing interests.
Figures



References
-
- Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol. 2017;13(1):37–51. - PubMed
