

This is a preprint.
Trans-Lesion Synthesis and Mismatch Repair Pathway Crosstalk Defines Chemoresistance and Hypermutation Mechanisms in Glioblastoma
- PMID: 37886584
- PMCID: PMC10602147
- DOI: 10.21203/rs.3.rs-2367368/v1
Trans-Lesion Synthesis and Mismatch Repair Pathway Crosstalk Defines Chemoresistance and Hypermutation Mechanisms in Glioblastoma
Update in
-
Trans-lesion synthesis and mismatch repair pathway crosstalk defines chemoresistance and hypermutation mechanisms in glioblastoma.Nat Commun. 2024 Mar 4;15(1):1957. doi: 10.1038/s41467-024-45979-5. Nat Commun. 2024. PMID: 38438348 Free PMC article.
Abstract
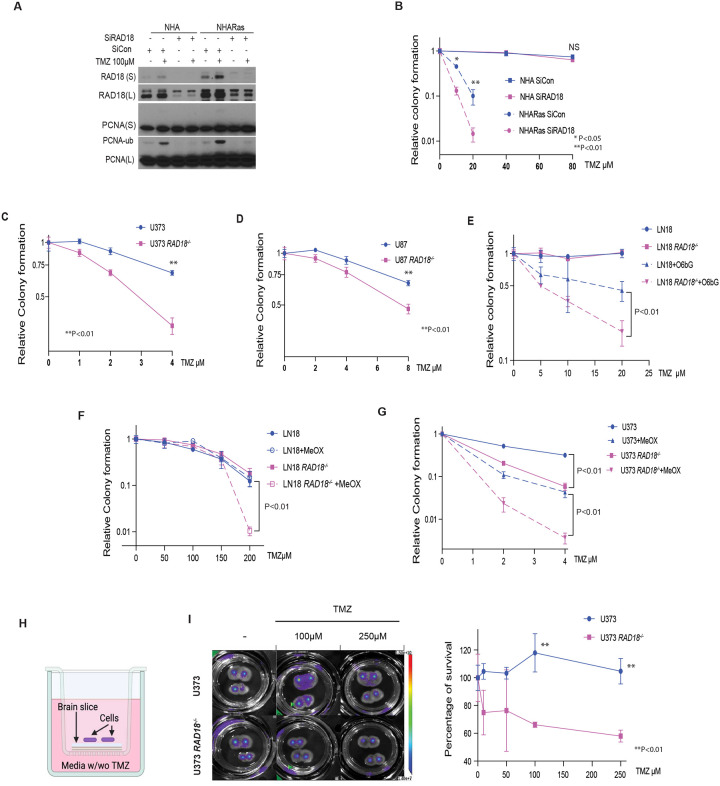
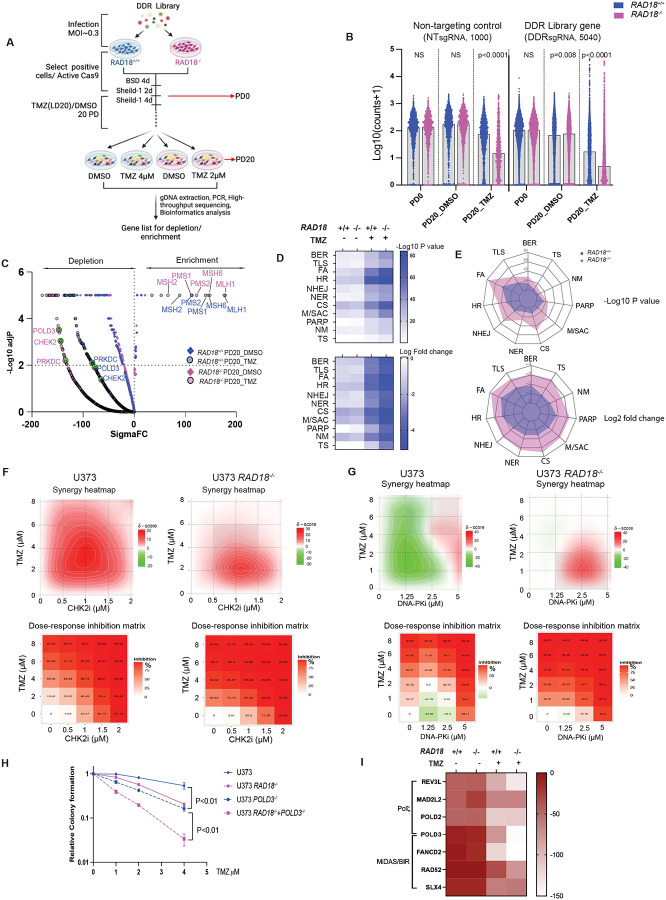
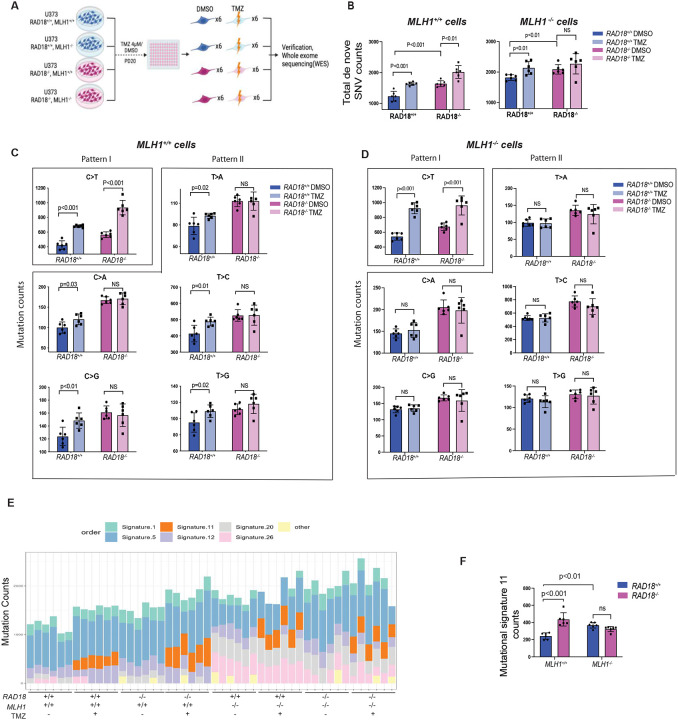
Almost all Glioblastoma (GBM) are either intrinsically resistant to the chemotherapeutical drug temozolomide (TMZ) or acquire therapy-induced mutations that cause chemoresistance and recurrence. The genome maintenance mechanisms responsible for GBM chemoresistance and hypermutation are unknown. We show that the E3 ubiquitin ligase RAD18 (a proximal regulator of TLS) is activated in a Mismatch repair (MMR)-dependent manner in TMZ-treated GBM cells, promoting post-replicative gap-filling and survival. An unbiased CRISPR screen provides a new aerial map of RAD18-interacting DNA damage response (DDR) pathways deployed by GBM to tolerate TMZ genotoxicity. Analysis of mutation signatures from TMZ-treated GBM reveals a role for RAD18 in error-free bypass of O6mG (the most toxic TMZ-induced lesion), and error-prone bypass of other TMZ-induced lesions. Our analyses of recurrent GBM patient samples establishes a correlation between low RAD18 expression and hypermutation. Taken together we define novel molecular underpinnings for the hallmark tumorigenic phenotypes of TMZ-treated GBM.
Conflict of interest statement
COMPETING INTERESTS DECLARATION B.M., S.H., and A.S. have submitted a provisional patent application based on the OBSC work in this manuscript
Figures




References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
