

Cooperativity and Frustration Effects (or Lack Thereof) in Polarizable and Non-polarizable Force Fields
- PMID: 37888874
- PMCID: PMC11078293
- DOI: 10.1021/acs.jctc.3c00762
Cooperativity and Frustration Effects (or Lack Thereof) in Polarizable and Non-polarizable Force Fields
Abstract
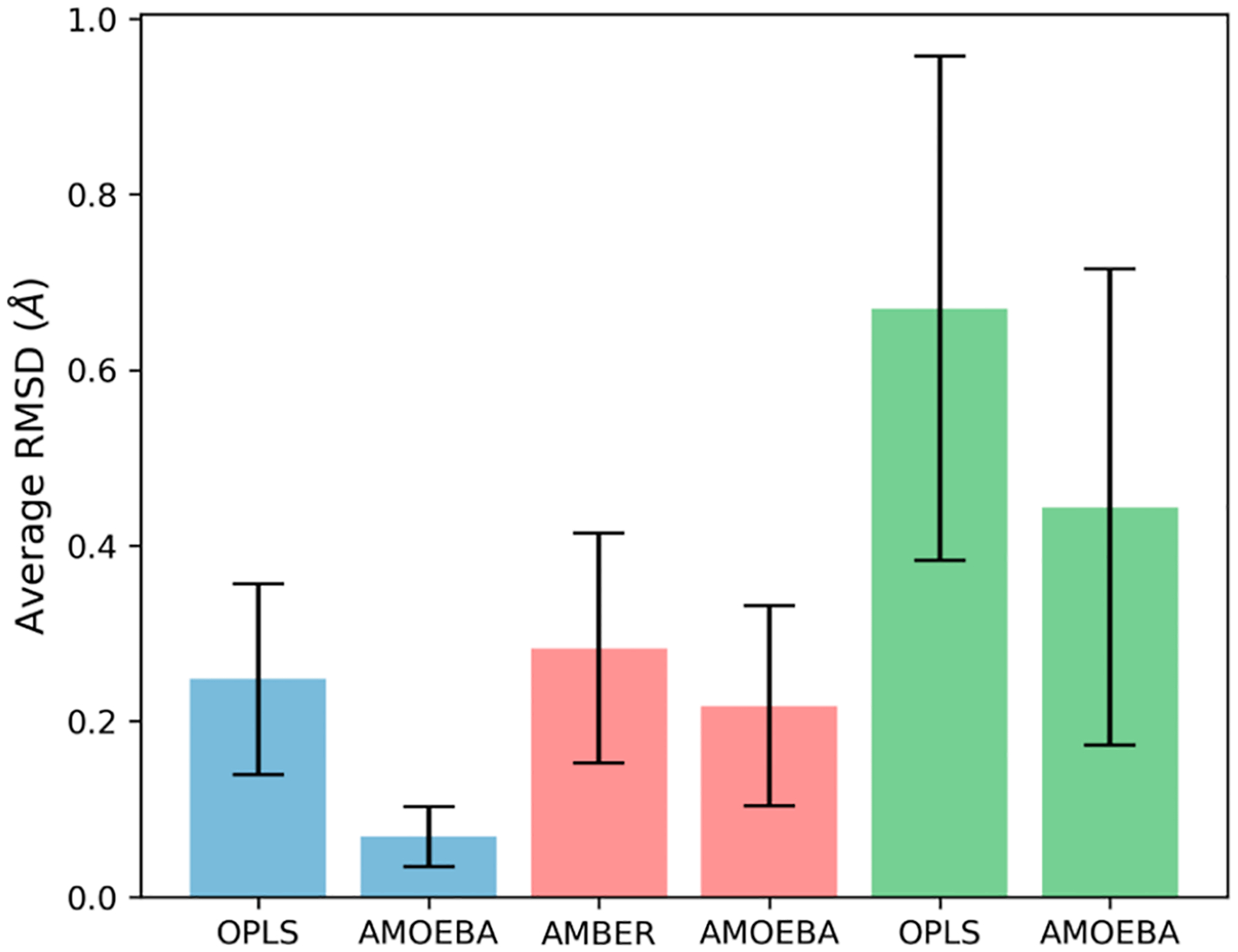
Understanding cooperativity and frustration is crucial for studying biological processes such as molecular recognition and protein aggregation. Force fields have been extensively utilized to explore cooperativity in the formation of protein secondary structures and self-assembled systems. Multiple studies have demonstrated that polarizable force fields provide more accurate descriptions of this phenomenon compared to fixed-charge pairwise nonpolarizable force fields, thanks to the incorporation of polarization effects. In this study, we assess the performance of the AMOEBA polarizable force field and the AMBER and OPLS nonpolarizable pairwise force fields in capturing positive and negative cooperativity recently explored in neutral and charged molecular clusters using density functional theory. Our findings show that polarizable and nonpolarizable force fields qualitatively reproduce the relative cooperativity observed in electron structure calculations. However, AMBER and OPLS fail to describe absolute cooperativity. In contrast, AMOEBA accounts for the absolute cooperativity by considering interactions beyond pairwise interactions. According to the energy decomposition analysis, it is observed that the electrostatic interactions calculated with the AMBER and OPLS force fields seem to play an important and counterintuitive role in reproducing the adiabatic interaction energies calculated with density functional theory. However, it is important to note that these force fields, due to their nature, do not explicitly incorporate many-body effects, which limits their ability to accurately describe cooperativity. On the other hand, frustration in polarizable and nonpolarizable force fields is caused by changes in bond stretching and angle bending terms of the building blocks when they are forming a complex.
Conflict of interest statement
The authors declare no competing financial interest.
Figures





References
-
- Haino T Cooperativity in molecular recognition of feet-to-feet-connected biscavitands. Pure Appl. Chem 2023, 95, 343–352.
-
- Parida S; Patra SK; Mishra S Self-Assembling Behaviour of Perylene, Perylene Diimide, and Thionated Perylene Diimide Deciphered through Non-Covalent Interactions. ChemPhysChem 2022, 23, No. e202200361. - PubMed
-
- Rudharachari Maiyelvaganan K; Prakash M; Kumar Ravva M Simultaneous interaction of graphene nanoflakes with cations and anions: A cooperativity study. Comput. Theor. Chem 2022, 1209, 113601.
-
- Liu S Principle of Chirality Hierarchy in Three-Blade Propeller Systems. J. Phys. Chem. Lett 2021, 12, 8720–8725. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources