

Nogo-B mediates endothelial oxidative stress and inflammation to promote coronary atherosclerosis in pressure-overloaded mouse hearts
- PMID: 37890359
- PMCID: PMC10633694
- DOI: 10.1016/j.redox.2023.102944
Nogo-B mediates endothelial oxidative stress and inflammation to promote coronary atherosclerosis in pressure-overloaded mouse hearts
Abstract
Aims: Endothelial dysfunction plays a pivotal role in atherosclerosis, but the detailed mechanism remains incomplete understood. Nogo-B is an endoplasmic reticulum (ER)-localized protein mediating ER-mitochondrial morphology. We previously showed endothelial Nogo-B as a key regulator of endothelial function in the setting of hypertension. Here, we aim to further assess the role of Nogo-B in coronary atherosclerosis in ApoE-/- mice with pressure overload.
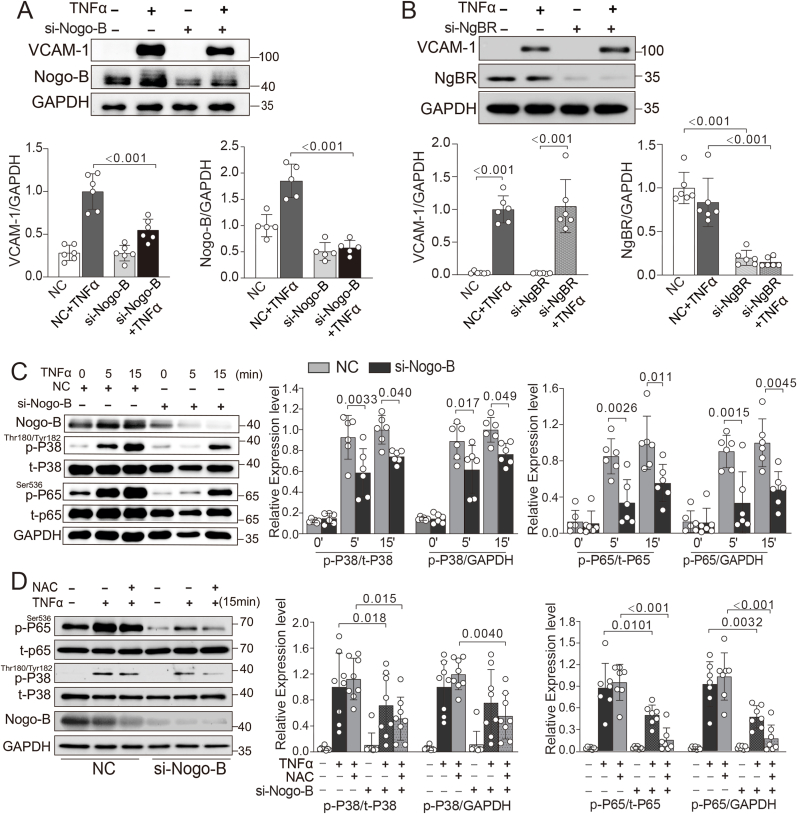
Methods and results: We generated double knockout (DKO) mouse models of systemically or endothelium-specifically excising Nogo-A/B gene on an ApoE-/- background. After 7 weeks of transverse aortic constriction (TAC) surgery, compared to ApoE-/- mice DKO mice were resistant to the development of coronary atherosclerotic lesions and plaque rapture. Sustained elevation of Nogo-B and adhesion molecules (VCAM-1/ICAM-1), early markers of atherosclerosis, was identified in heart tissues and endothelial cells (ECs) isolated from TAC ApoE-/- mice, changes that were significantly repressed by Nogo-B deficiency. In cultured human umbilical vein endothelial cells (HUVECs) exposure to inflammatory cytokines (TNF-α, IL-1β), Nogo-B was upregulated and activated reactive oxide species (ROS)-p38-p65 signaling axis. Mitofusin 2 (Mfn2) is a key protein tethering ER to mitochondria in ECs, and we showed that Nogo-B expression positively correlated with Mfn2 protein level. And Nogo-B deletion in ECs or in ApoE-/- mice reduced Mfn2 protein content and increased ER-mitochondria distance, reduced ER-mitochondrial Ca2+ transport and mitochondrial ROS generation, and prevented VCAM-1/ICAM-1 upregulation and EC dysfunction, eventually restrained atherosclerotic lesions development.
Conclusion: Our study revealed that Nogo-B is a critical modulator in promoting endothelial dysfunction and consequent pathogenesis of coronary atherosclerosis in pressure overloaded hearts of ApoE-/- mice. Nogo-B may hold the promise to be a common therapeutic target in the setting of hypertension.
Keywords: Coronary atherosclerosis; Mitochondria; Nogo-B; Pressure overload; Reactive oxygen species.
Copyright © 2023 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that there are no confilct of interests.
Figures








References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
