

CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico
- PMID: 37896794
- PMCID: PMC10611021
- DOI: 10.3390/v15102017
CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico
Abstract
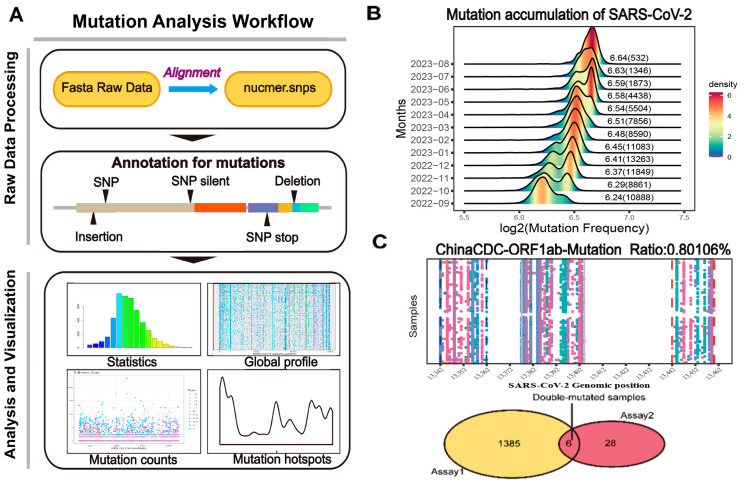
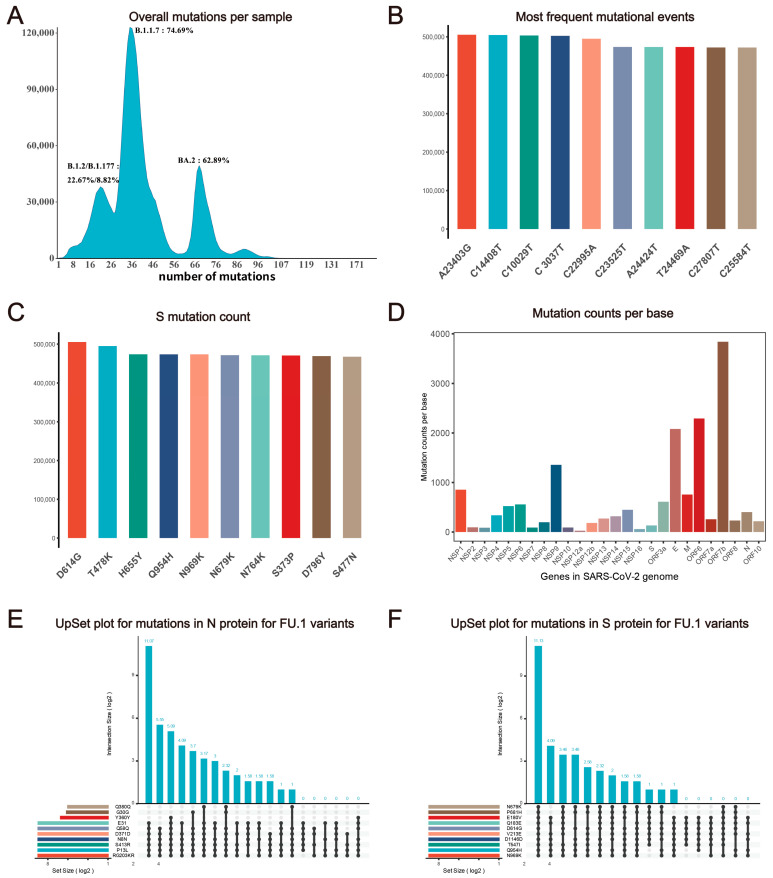
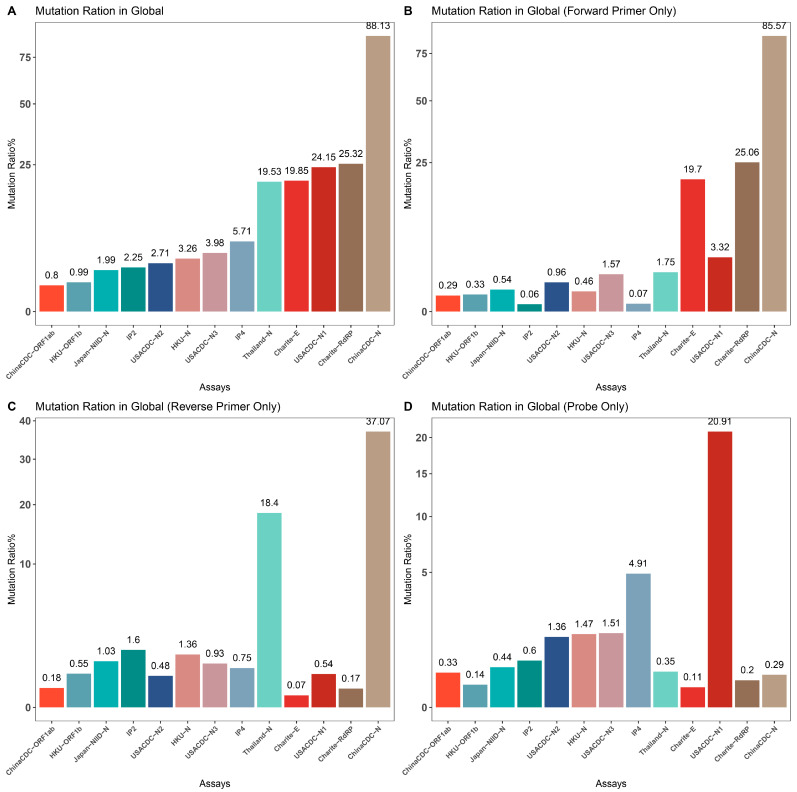
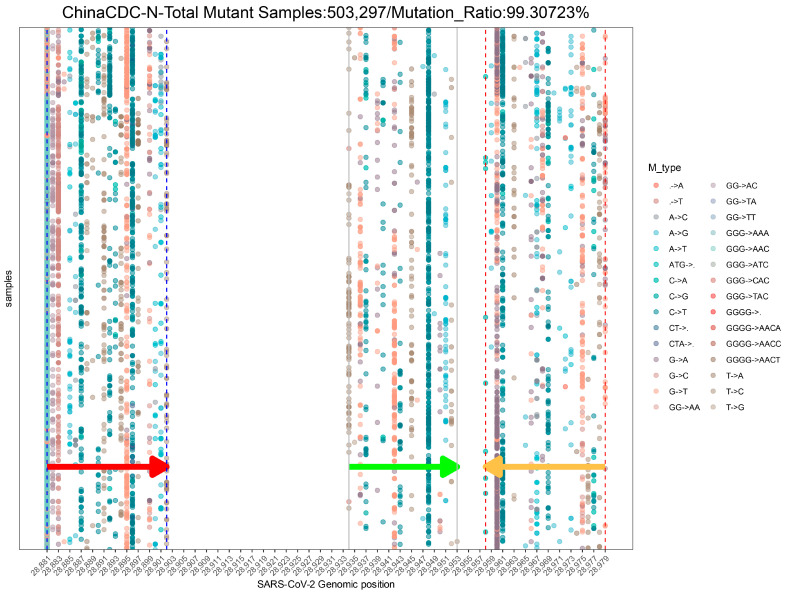
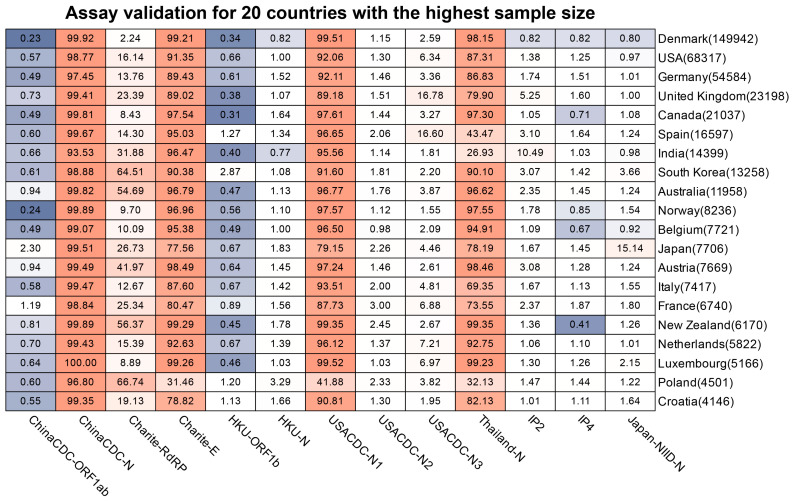
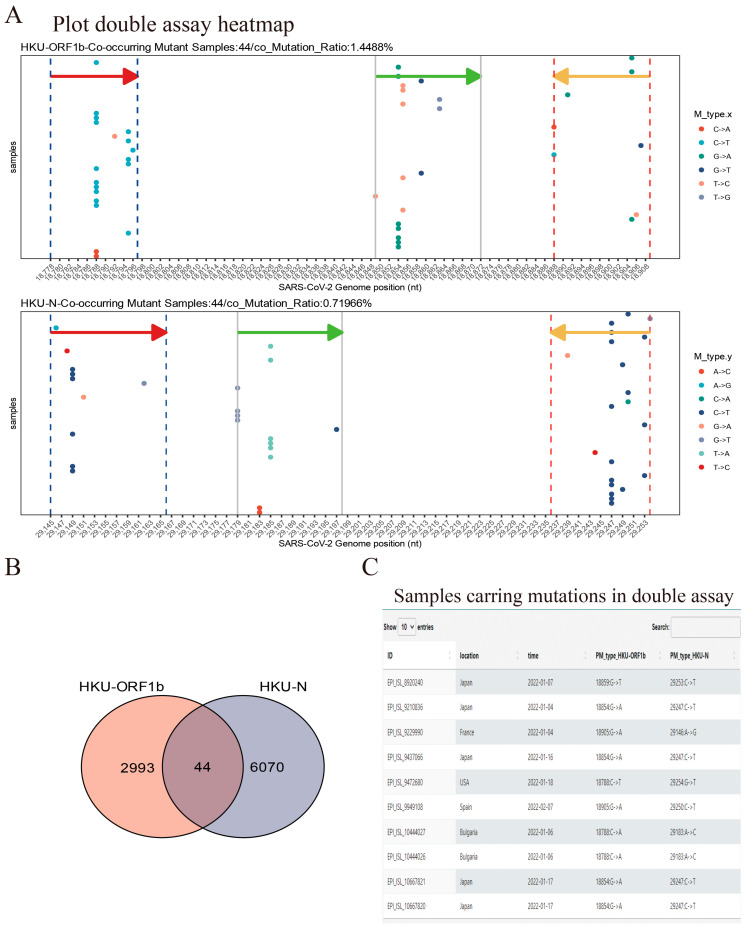
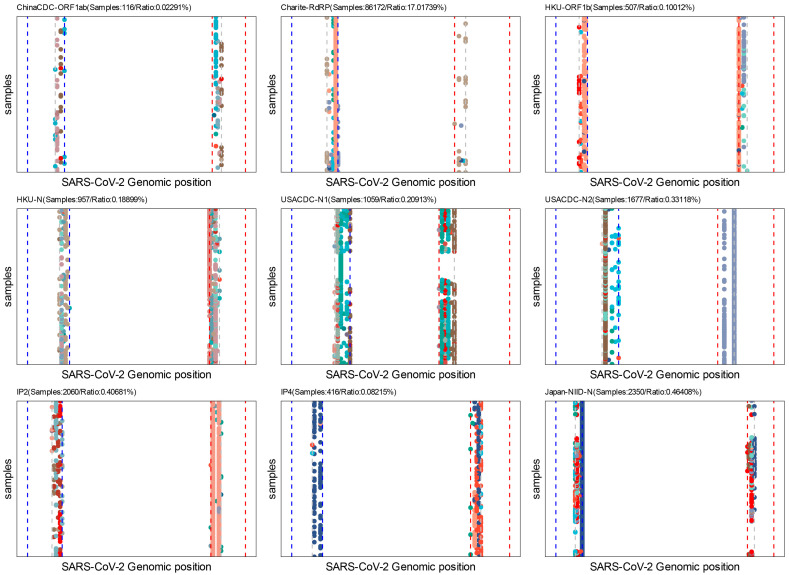
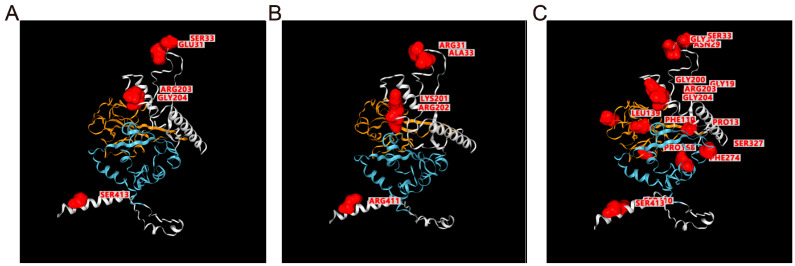
The coronavirus disease 2019 (COVID-19) pandemic is still ongoing, with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continuing to evolve and accumulate mutations. While various bioinformatics tools have been developed for SARS-CoV-2, a well-curated mutation-tracking database integrated with in silico evaluation for molecular diagnostic assays is currently unavailable. To address this, we introduce CovidShiny, a web tool that integrates mutation profiling, in silico evaluation, and data download capabilities for genomic sequence-based SARS-CoV-2 assays and data download. It offers a feasible framework for surveilling the mutation of SARS-CoV-2 and evaluating the coverage of the molecular diagnostic assay for SARS-CoV-2. With CovidShiny, we examined the dynamic mutation pattern of SARS-CoV-2 and evaluated the coverage of commonly used assays on a large scale. Based on our in silico analysis, we stress the importance of using multiple target molecular diagnostic assays for SARS-CoV-2 to avoid potential false-negative results caused by viral mutations. Overall, CovidShiny is a valuable tool for SARS-CoV-2 mutation surveillance and in silico assay design and evaluation.
Keywords: SARS-CoV-2; assay evaluation; integrated web tool; mutation analysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Design and Implementation of Improved SARS-CoV-2 Diagnostic Assays To Mitigate the Impact of Genomic Mutations on Target Failure: the Xpert Xpress SARS-CoV-2 Experience.Microbiol Spectr. 2022 Dec 21;10(6):e0135522. doi: 10.1128/spectrum.01355-22. Epub 2022 Oct 18. Microbiol Spectr. 2022. PMID: 36255326 Free PMC article.
-
Deepening of In Silico Evaluation of SARS-CoV-2 Detection RT-qPCR Assays in the Context of New Variants.Genes (Basel). 2021 Apr 13;12(4):565. doi: 10.3390/genes12040565. Genes (Basel). 2021. PMID: 33924636 Free PMC article.
-
In silico evaluation of the impact of Omicron variant of concern sublineage BA.4 and BA.5 on the sensitivity of RT-qPCR assays for SARS-CoV-2 detection using whole genome sequencing.J Med Virol. 2023 Jan;95(1):e28241. doi: 10.1002/jmv.28241. Epub 2022 Nov 8. J Med Virol. 2023. PMID: 36263448 Free PMC article.
-
[SARS-CoV-2 and Microbiological Diagnostic Dynamics in COVID-19 Pandemic].Mikrobiyol Bul. 2020 Jul;54(3):497-509. doi: 10.5578/mb.69839. Mikrobiyol Bul. 2020. PMID: 32755524 Review. Turkish.
-
Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic.Int J Mol Sci. 2021 Jan 28;22(3):1298. doi: 10.3390/ijms22031298. Int J Mol Sci. 2021. PMID: 33525651 Free PMC article. Review.
References
-
- WHO Coronavirus (COVID-19) Dashboard. [(accessed on 18 July 2023)]. Available online: https://covid19.who.int/
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
