

Pathogenesis and therapeutic implications of EBV-associated epithelial cancers
- PMID: 37901329
- PMCID: PMC10600384
- DOI: 10.3389/fonc.2023.1202117
Pathogenesis and therapeutic implications of EBV-associated epithelial cancers
Abstract
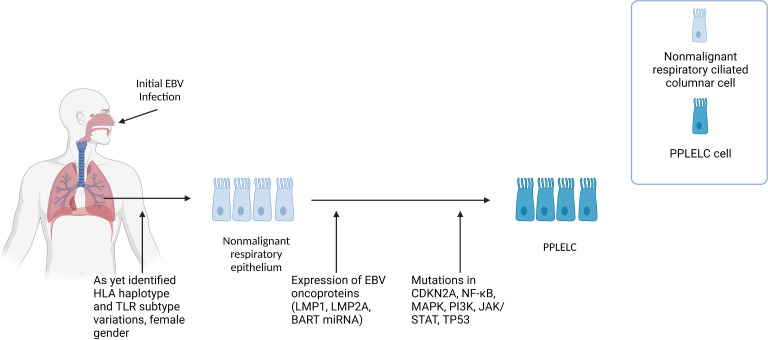
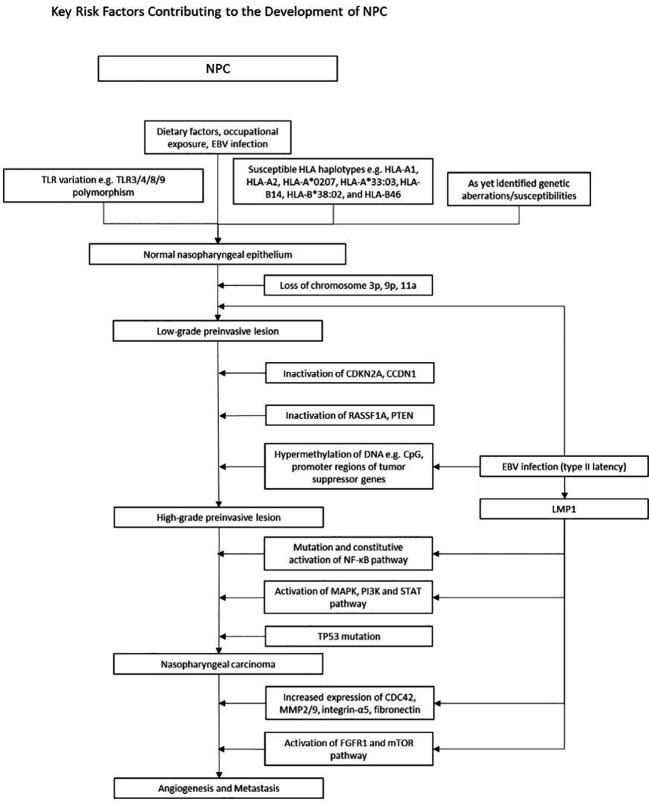
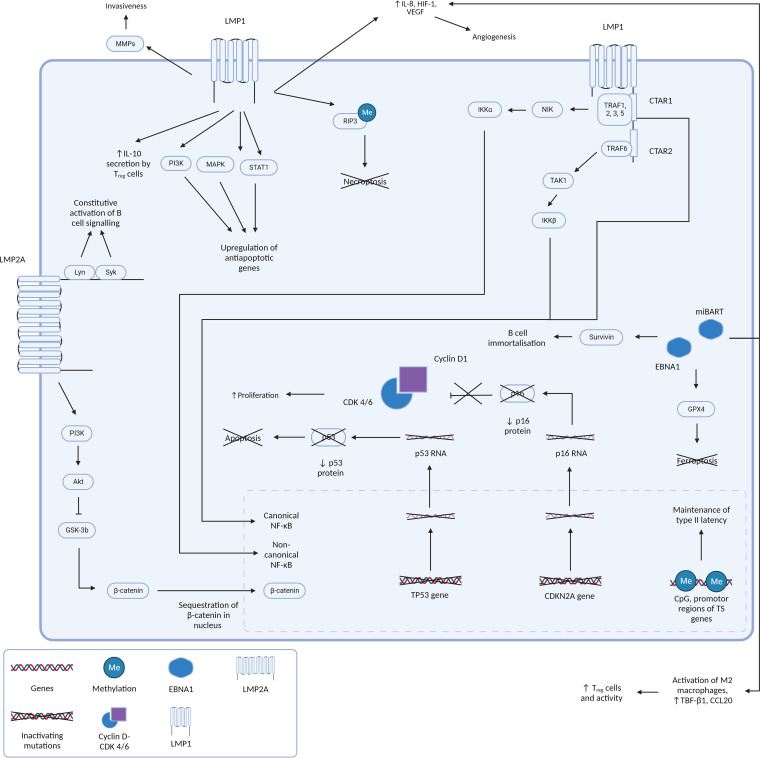
Epstein-Barr virus (EBV), one of the most common human viruses, has been associated with both lymphoid and epithelial cancers. Undifferentiated nasopharyngeal carcinoma (NPC), EBV associated gastric cancer (EBVaGC) and lymphoepithelioma-like carcinoma (LELC) are amongst the few common epithelial cancers that EBV has been associated with. The pathogenesis of EBV-associated NPC has been well described, however, the same cannot be said for primary pulmonary LELC (PPLELC) owing to the rarity of the cancer. In this review, we outline the pathogenesis of EBV-associated NPC and EBVaGCs and their recent advances. By drawing on similarities between NPC and PPLELC, we then also postulated the pathogenesis of PPLELC. A deeper understanding about the pathogenesis of EBV enables us to postulate the pathogenesis of other EBV associated cancers such as PPLELC.
Keywords: Epstein-Barr virus; lymphoepithelioma-like carcinoma; nasopharyngeal cancer; pathogenesis; primary pulmonary lymphoepithelioma-like carcinoma.
Copyright © 2023 Low, Loh, Peh, Chu, Han and Toh.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Epstein-Barr Virus Epithelial Cancers-A Comprehensive Understanding to Drive Novel Therapies.Front Immunol. 2021 Dec 10;12:734293. doi: 10.3389/fimmu.2021.734293. eCollection 2021. Front Immunol. 2021. PMID: 34956172 Free PMC article. Review.
-
Epstein-Barr Virus-Induced Epigenetic Pathogenesis of Viral-Associated Lymphoepithelioma-Like Carcinomas and Natural Killer/T-Cell Lymphomas.Pathogens. 2018 Jul 18;7(3):63. doi: 10.3390/pathogens7030063. Pathogens. 2018. PMID: 30022006 Free PMC article. Review.
-
Epstein-Barr virus infection and nasopharyngeal carcinoma.Philos Trans R Soc Lond B Biol Sci. 2017 Oct 19;372(1732):20160270. doi: 10.1098/rstb.2016.0270. Philos Trans R Soc Lond B Biol Sci. 2017. PMID: 28893937 Free PMC article. Review.
-
Is gastric lymphoepithelioma-like carcinoma a special subtype of EBV-associated gastric carcinoma? New insight based on clinicopathological features and EBV genome polymorphisms.Gastric Cancer. 2015 Apr;18(2):246-55. doi: 10.1007/s10120-014-0376-9. Epub 2014 Apr 27. Gastric Cancer. 2015. PMID: 24771002
-
Oncogenic induction of cellular high CpG methylation by Epstein-Barr virus in malignant epithelial cells.Chin J Cancer. 2014 Dec;33(12):604-8. doi: 10.5732/cjc.014.10191. Epub 2014 Oct 17. Chin J Cancer. 2014. PMID: 25322866 Free PMC article.
Cited by
-
Viral oncogenesis in cancer: from mechanisms to therapeutics.Signal Transduct Target Ther. 2025 May 12;10(1):151. doi: 10.1038/s41392-025-02197-9. Signal Transduct Target Ther. 2025. PMID: 40350456 Free PMC article. Review.
-
Precision Medicine for Nasopharyngeal Cancer-A Review of Current Prognostic Strategies.Cancers (Basel). 2024 Feb 24;16(5):918. doi: 10.3390/cancers16050918. Cancers (Basel). 2024. PMID: 38473280 Free PMC article. Review.
-
Contrast-enhanced CT and PET-CT characteristics of primary tracheal lymphoepithelioma-like carcinoma: case series.Transl Lung Cancer Res. 2024 May 31;13(5):1101-1109. doi: 10.21037/tlcr-24-333. Epub 2024 May 29. Transl Lung Cancer Res. 2024. PMID: 38854950 Free PMC article.
-
Association between Epstein Barr virus and Oral Lichen Planus clinical phenotypes and p53 expression.Sci Rep. 2025 Jul 24;15(1):26977. doi: 10.1038/s41598-025-12095-3. Sci Rep. 2025. PMID: 40707534 Free PMC article.
-
Epigenetic and epitranscriptomic regulation during oncogenic γ-herpesvirus infection.Front Microbiol. 2025 Jan 7;15:1484455. doi: 10.3389/fmicb.2024.1484455. eCollection 2024. Front Microbiol. 2025. PMID: 39839102 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources