

The CRL3gigaxonin ubiquitin ligase-USP15 pathway governs the destruction of neurofilament proteins
- PMID: 37903270
- PMCID: PMC10636361
- DOI: 10.1073/pnas.2306395120
The CRL3gigaxonin ubiquitin ligase-USP15 pathway governs the destruction of neurofilament proteins
Abstract
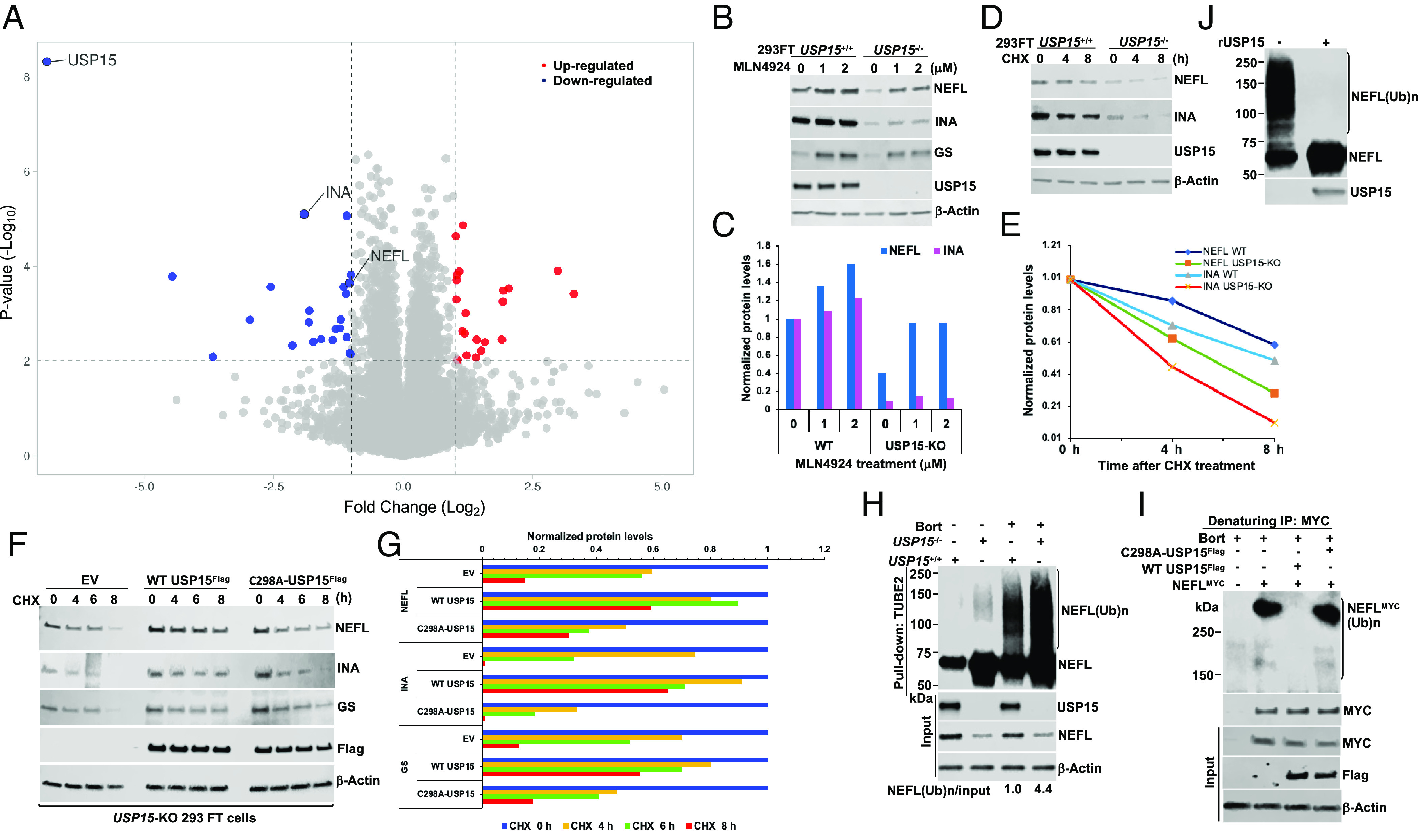
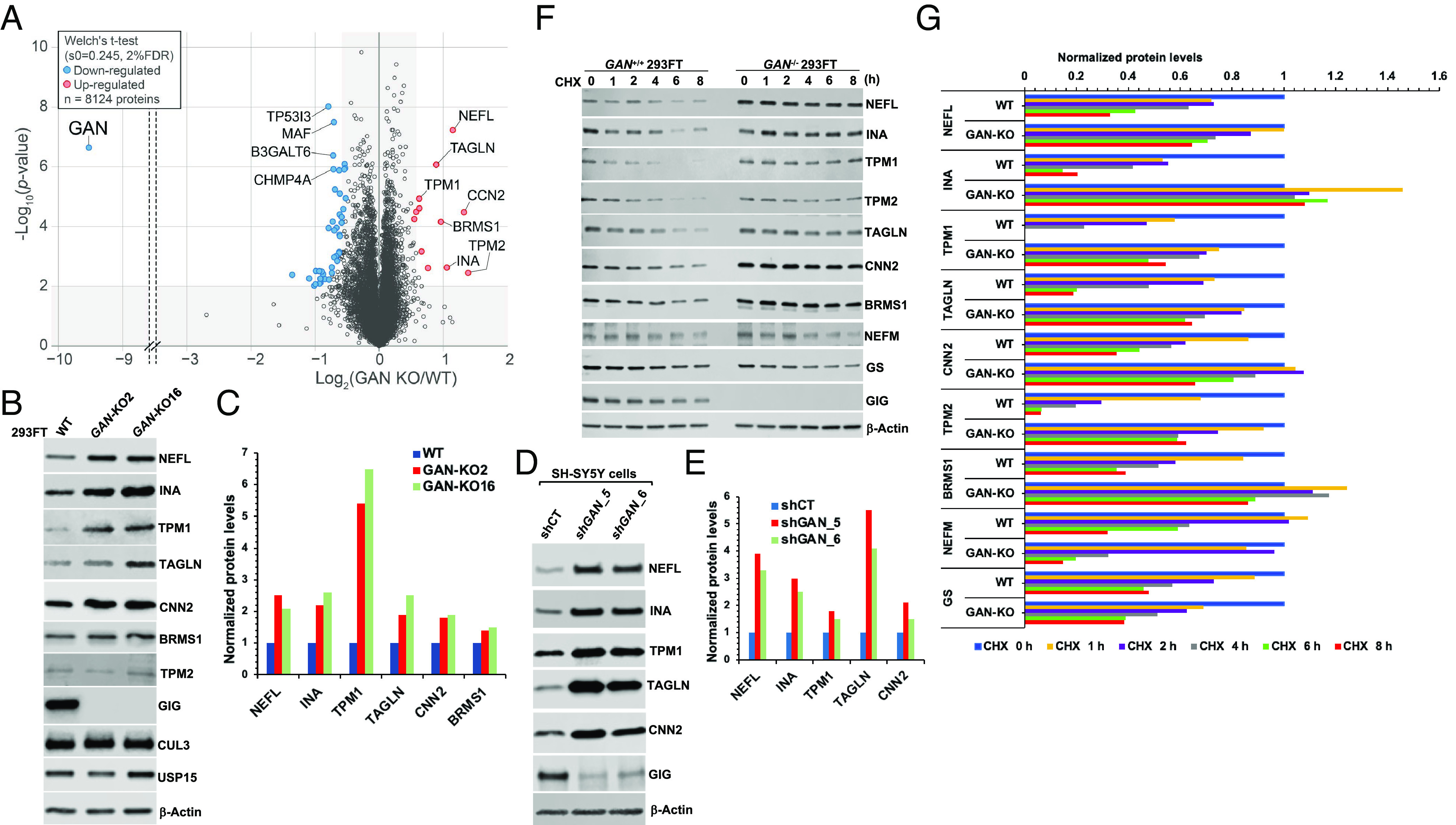
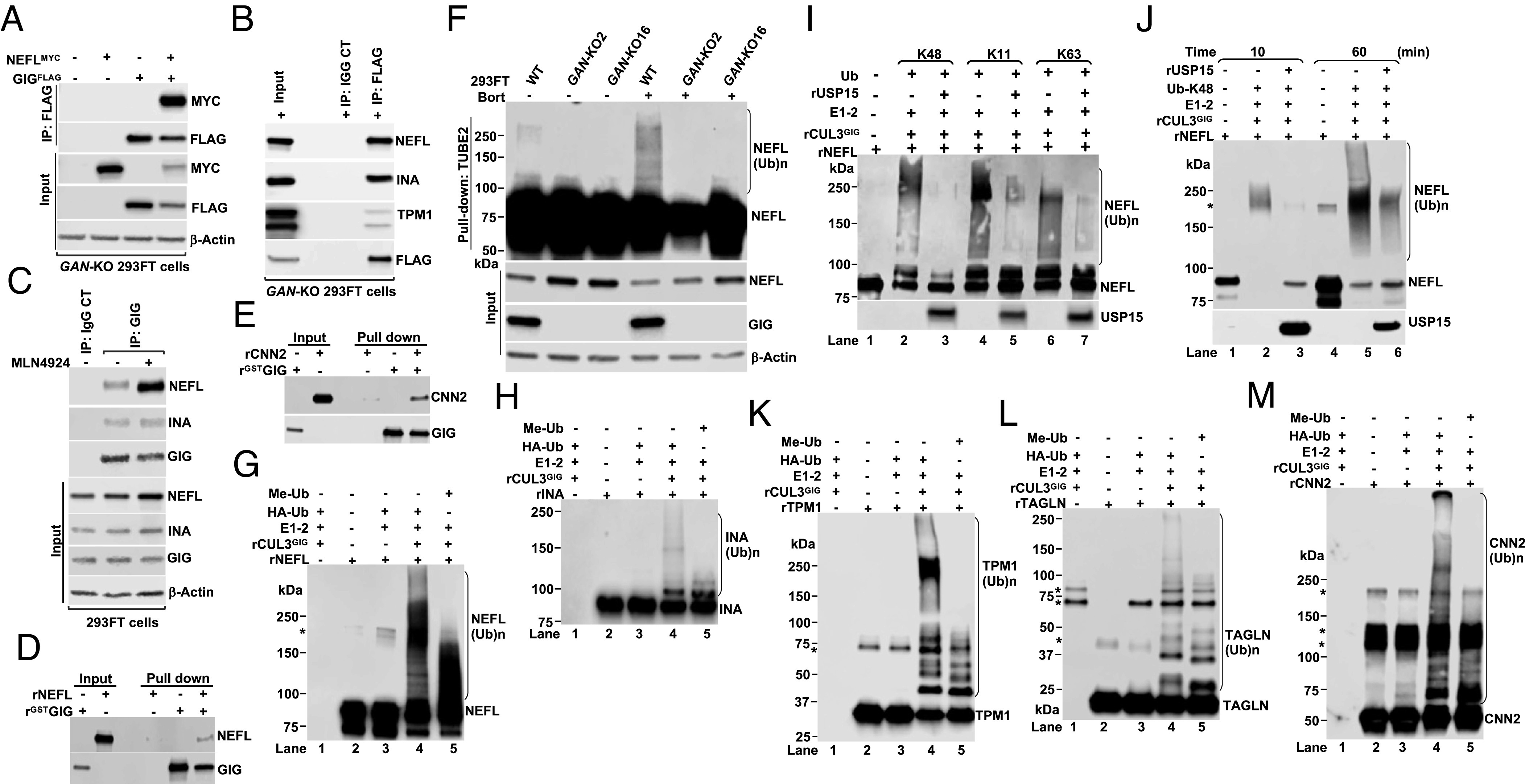
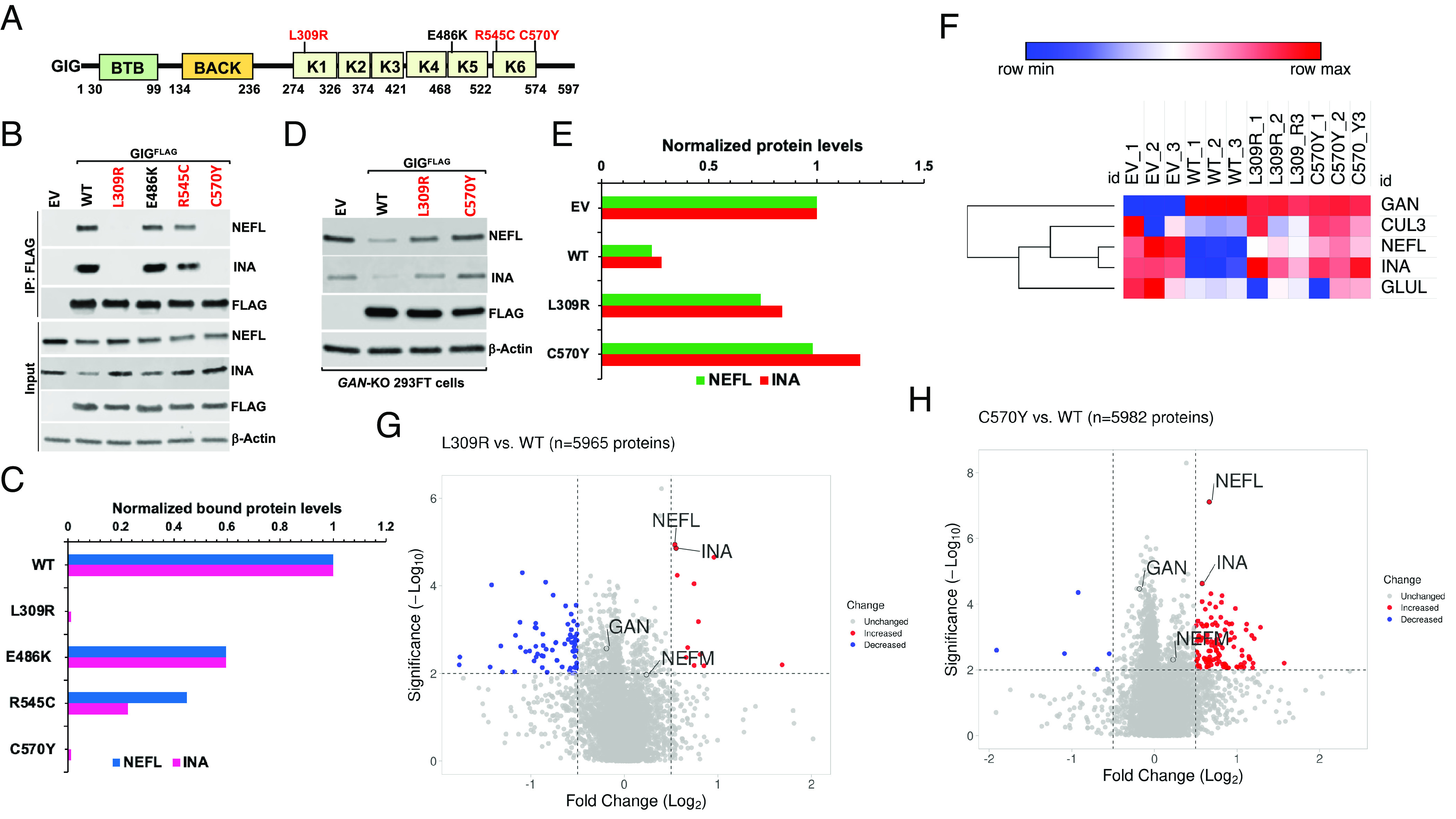
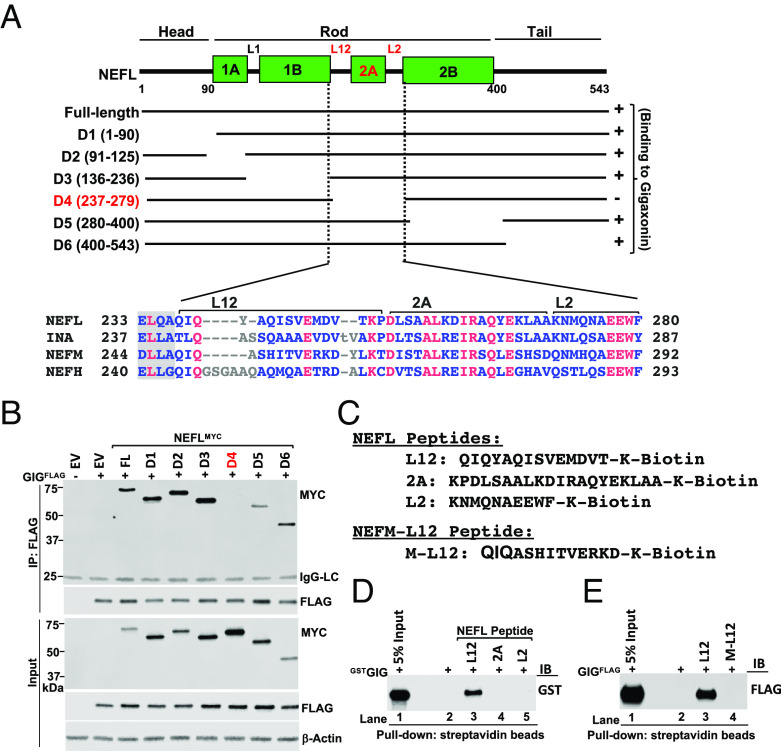
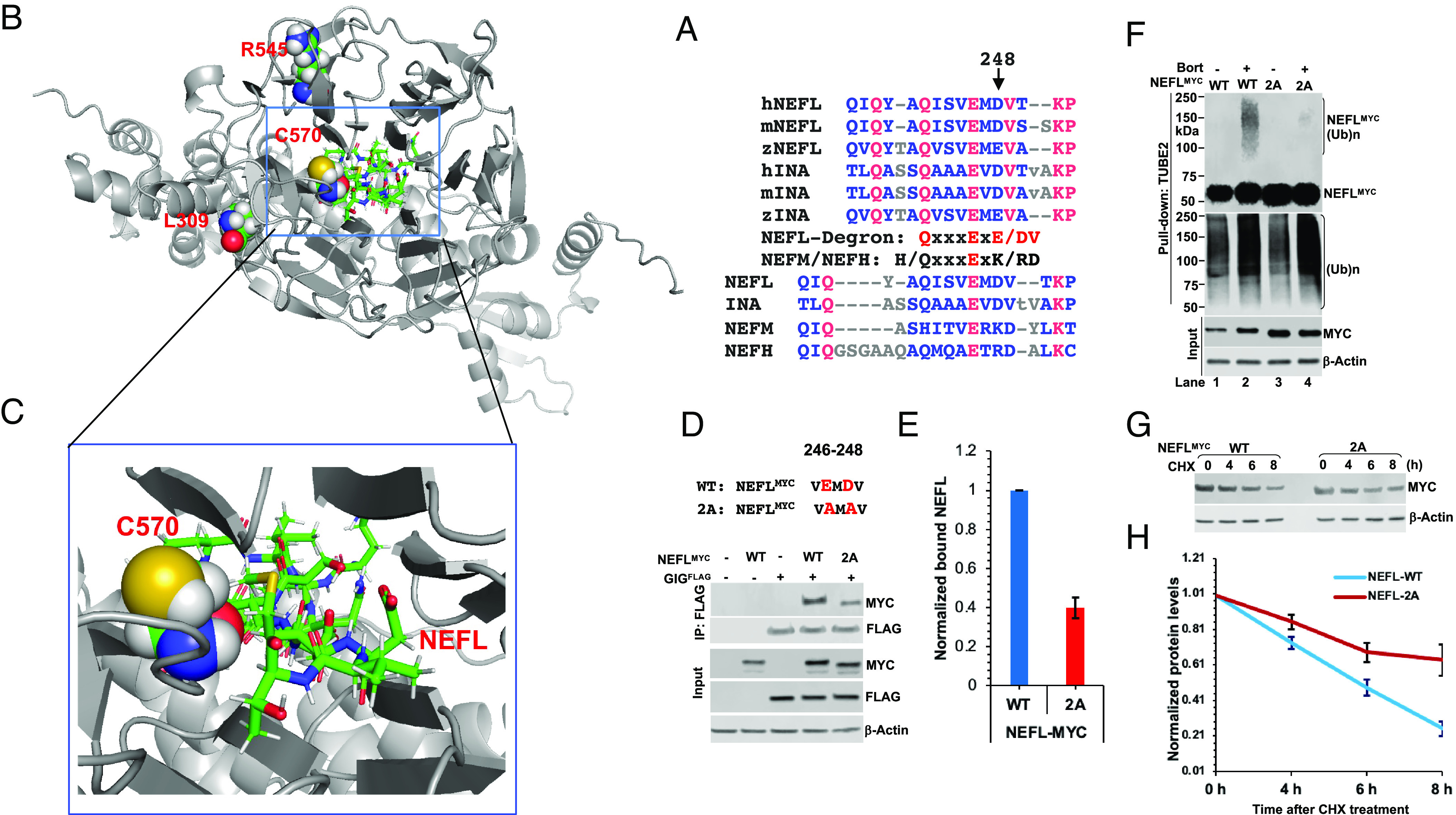
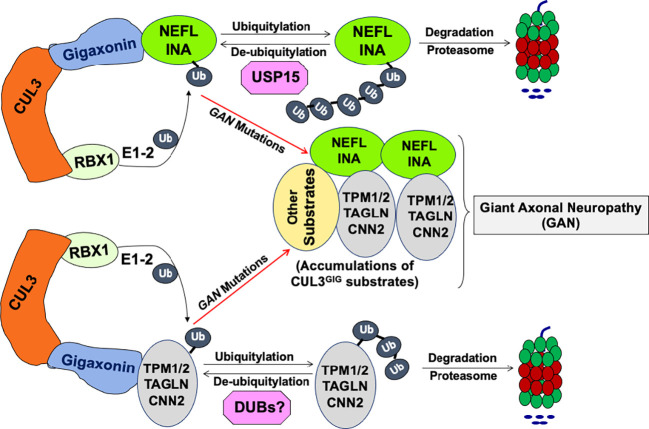
Giant axonal neuropathy (GAN) is caused by mutations in the GAN gene encoding for gigaxonin (GIG), which functions as an adaptor of the CUL3-RBX1-GIG (CRL3GIG) E3 ubiquitin ligase complex. The pathological hallmark of GAN is characterized by the accumulation of densely packed neurofilaments (NFs) in the axons. However, there are fundamental knowledge gaps in our understanding of the molecular mechanisms by which the ubiquitin-proteasome system controls the homeostasis of NF proteins. Recently, the deubiquitylating enzyme USP15 was reported to play a crucial role in regulating ubiquitylation and proteasomal degradation of CRL4CRBN substrate proteins. Here, we report that the CRL3GIG-USP15 pathway governs the destruction of NF proteins NEFL and INA. We identified a specific degron called NEFLL12 degron for CRL3GIG. Notably, mutations in the C-terminal Kelch domain of GIG, represented by L309R, R545C, and C570Y, disrupted the binding of GIG to NEFL and INA, leading to the accumulation of these NF proteins. This accounts for the loss-of-function mutations in GAN patients. In addition to regulating NFs, CRL3GIG also controls actin filaments by directly targeting actin-filament-binding regulatory proteins TPM1, TPM2, TAGLN, and CNN2 for proteasomal degradation. Thus, our findings broadly impact the field by providing fundamental mechanistic insights into regulating extremely long-lived NF proteins NEFL and INA by the CRL3GIG-USP15 pathway and offering previously unexplored therapeutic opportunities to treat GAN patients and other neurodegenerative diseases by explicitly targeting downstream substrates of CRL3GIG.
Keywords: GAN; Gigaxonin; USP15; neurofilament.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Varshavsky A., The ubiquitin system, an immense realm. Annu. Rev. Biochem. 81, 167–176 (2012). - PubMed
-
- Hershko A., Ciechanover A., The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998). - PubMed
-
- Petroski M. D., Deshaies R. J., Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
