

Isoform-resolved transcriptome of the human preimplantation embryo
- PMID: 37903791
- PMCID: PMC10616205
- DOI: 10.1038/s41467-023-42558-y
Isoform-resolved transcriptome of the human preimplantation embryo
Abstract
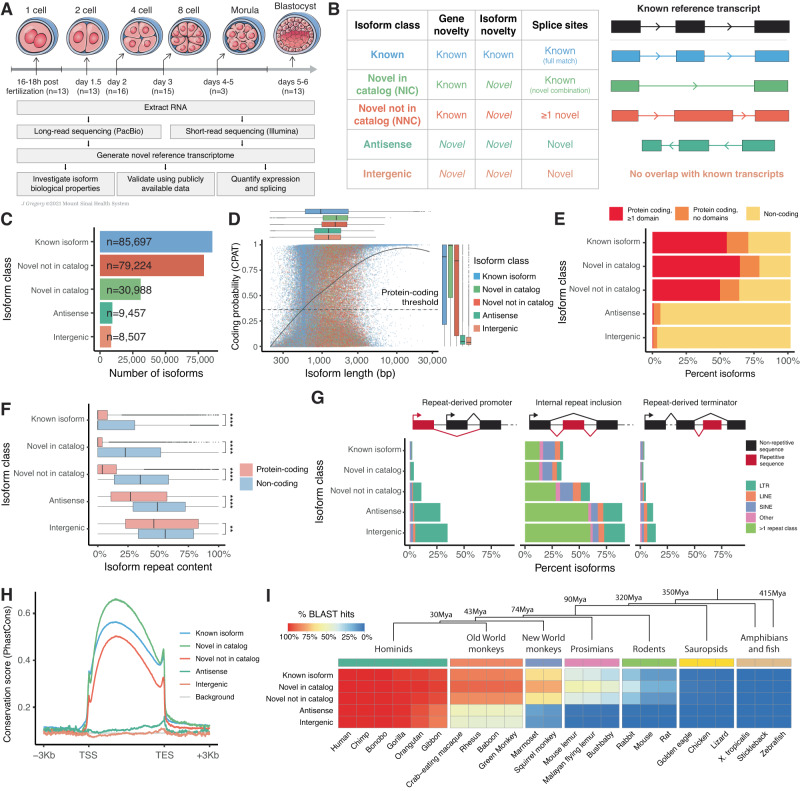
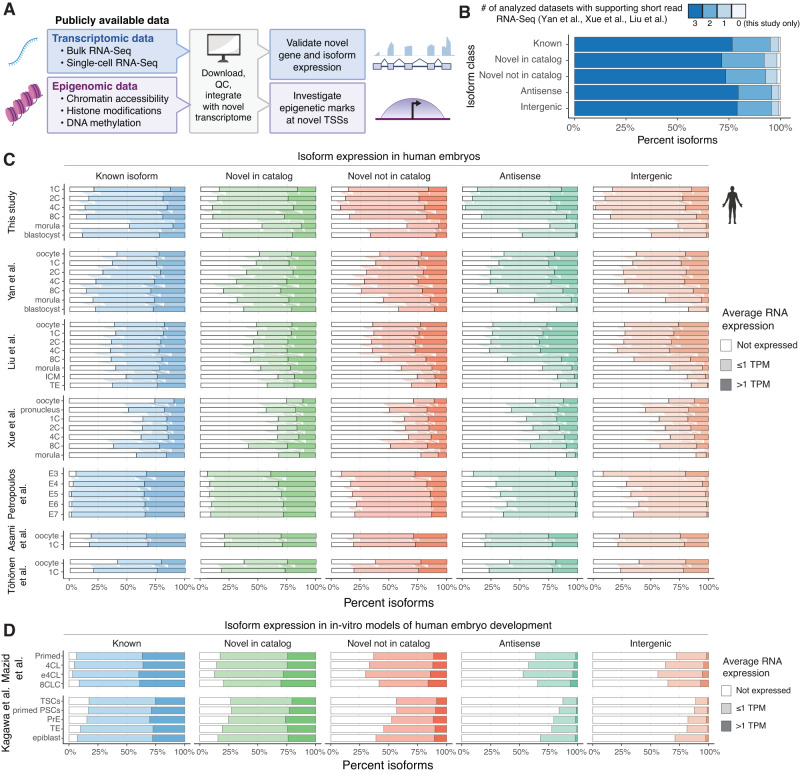
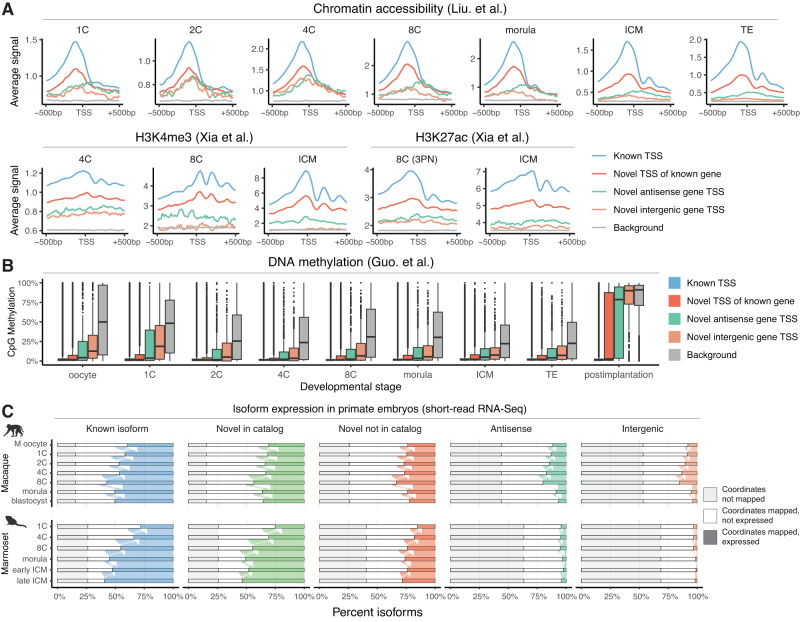
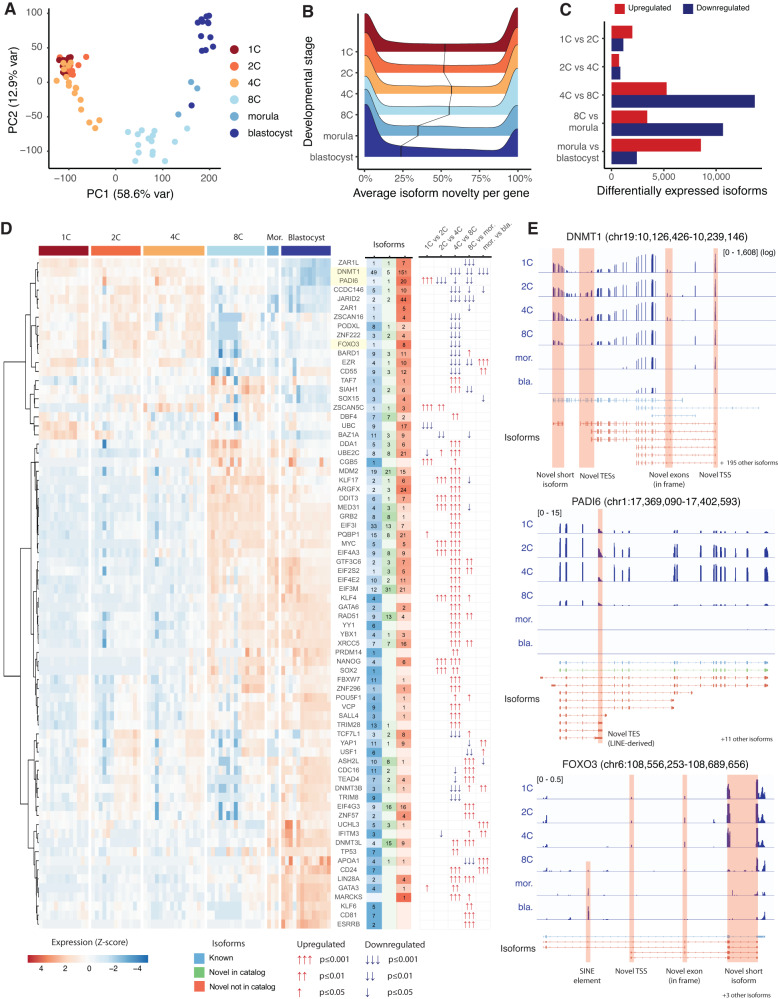
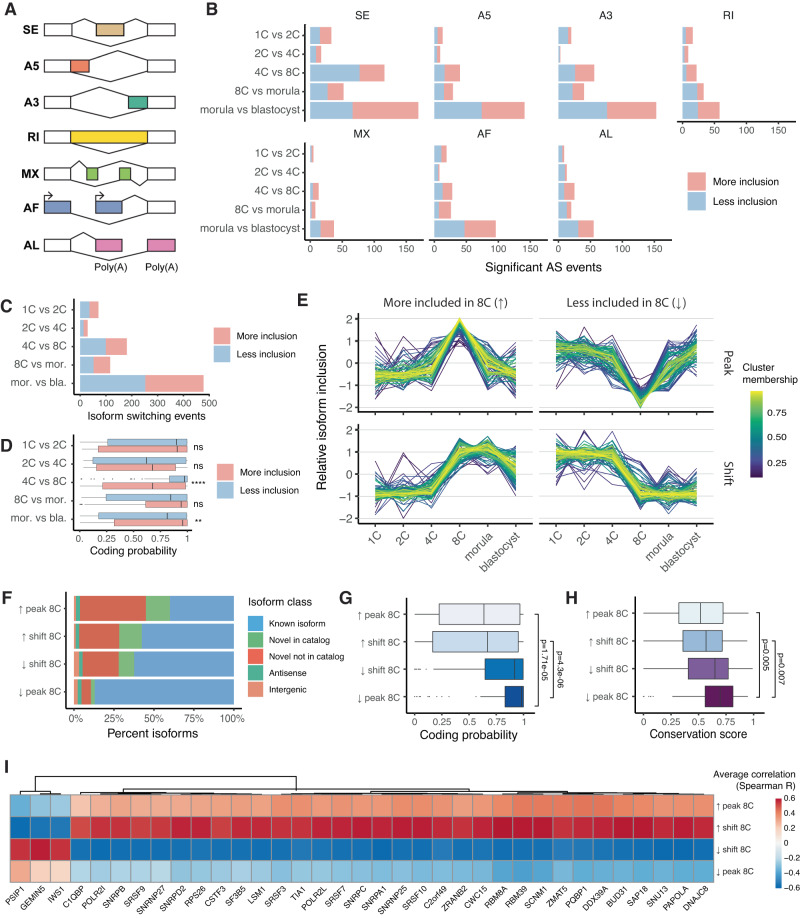
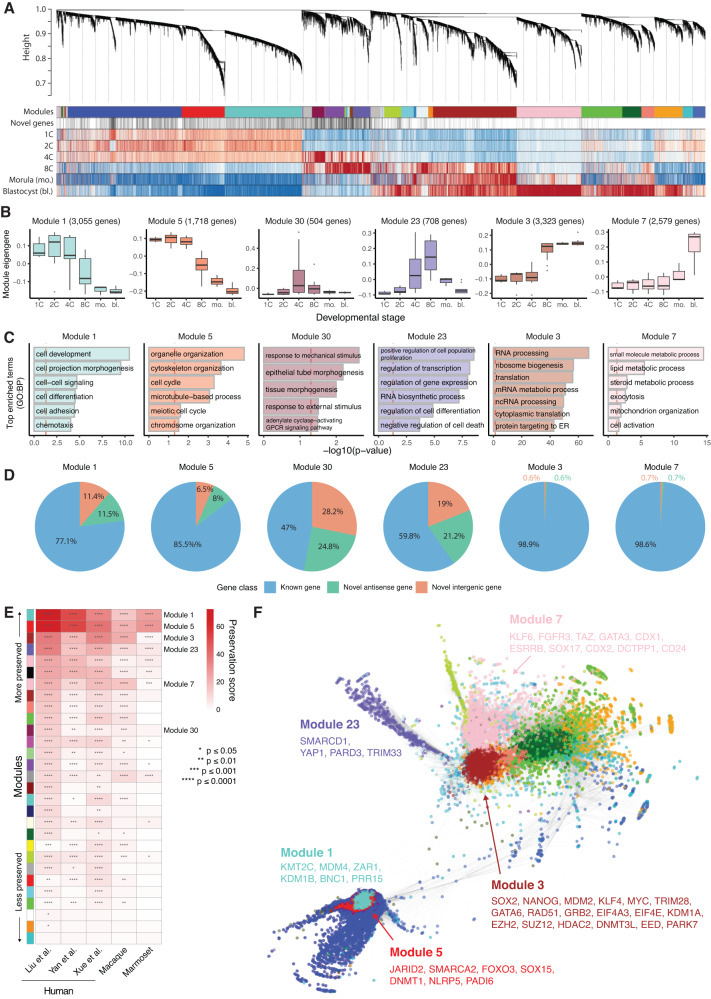
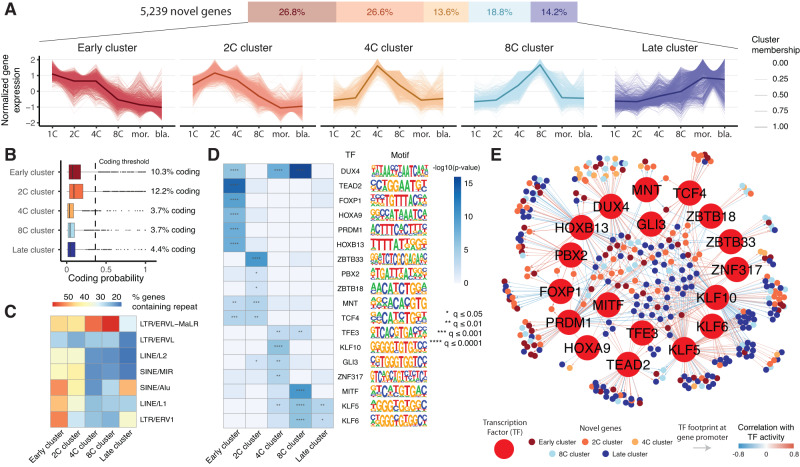
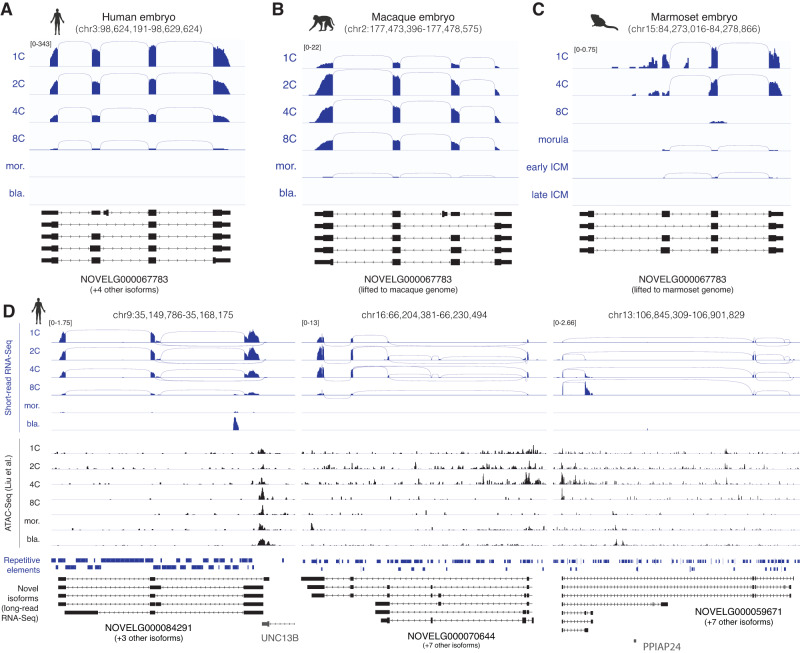
Human preimplantation development involves extensive remodeling of RNA expression and splicing. However, its transcriptome has been compiled using short-read sequencing data, which fails to capture most full-length mRNAs. Here, we generate an isoform-resolved transcriptome of early human development by performing long- and short-read RNA sequencing on 73 embryos spanning the zygote to blastocyst stages. We identify 110,212 unannotated isoforms transcribed from known genes, including highly conserved protein-coding loci and key developmental regulators. We further identify 17,964 isoforms from 5,239 unannotated genes, which are largely non-coding, primate-specific, and highly associated with transposable elements. These isoforms are widely supported by the integration of published multi-omics datasets, including single-cell 8CLC and blastoid studies. Alternative splicing and gene co-expression network analyses further reveal that embryonic genome activation is associated with splicing disruption and transient upregulation of gene modules. Together, these findings show that the human embryo transcriptome is far more complex than currently known, and will act as a valuable resource to empower future studies exploring development.
© 2023. The Author(s).
Conflict of interest statement
R.P.S. is an equity holder and paid consultant to GeneDx. However, the research was solely conducted by MSSM and TASMC facilities. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
