

Proteomics reveal biomarkers for diagnosis, disease activity and long-term disability outcomes in multiple sclerosis
- PMID: 37903821
- PMCID: PMC10616092
- DOI: 10.1038/s41467-023-42682-9
Proteomics reveal biomarkers for diagnosis, disease activity and long-term disability outcomes in multiple sclerosis
Abstract
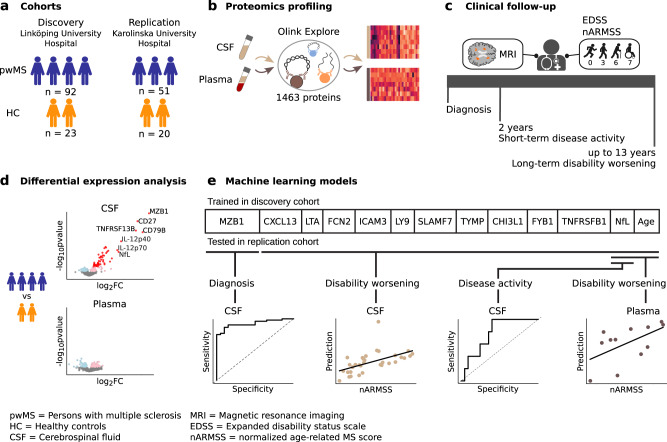
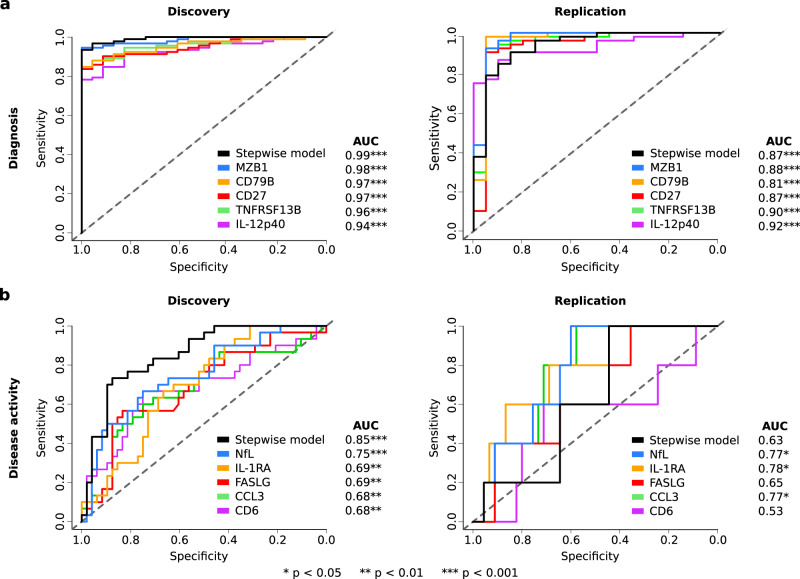
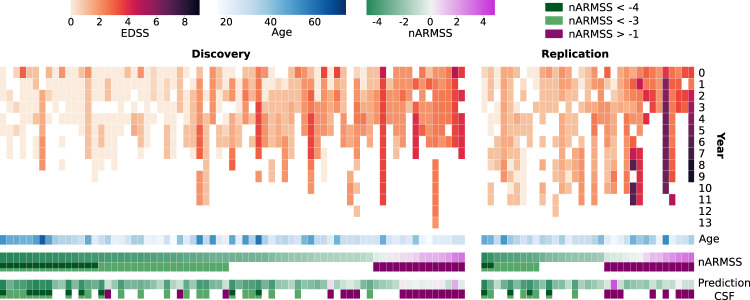
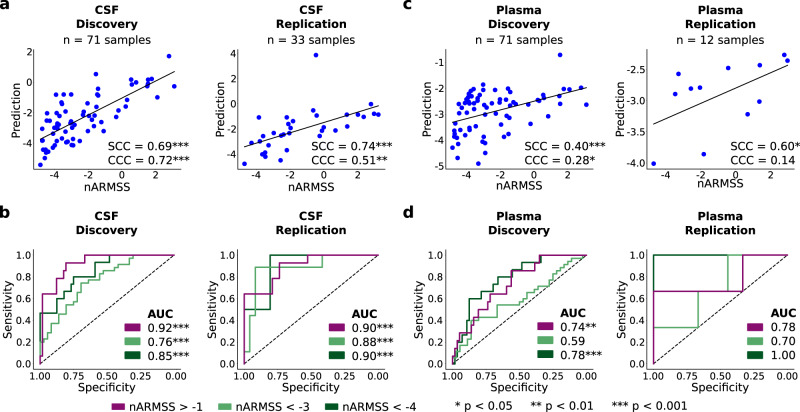
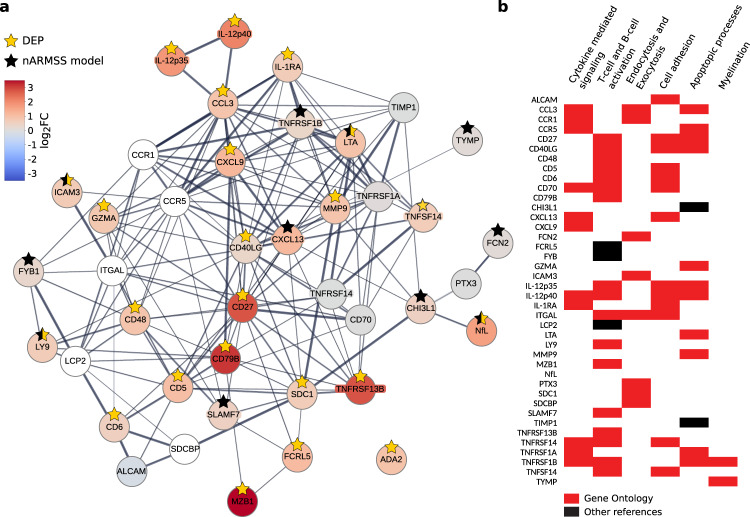
Sensitive and reliable protein biomarkers are needed to predict disease trajectory and personalize treatment strategies for multiple sclerosis (MS). Here, we use the highly sensitive proximity-extension assay combined with next-generation sequencing (Olink Explore) to quantify 1463 proteins in cerebrospinal fluid (CSF) and plasma from 143 people with early-stage MS and 43 healthy controls. With longitudinally followed discovery and replication cohorts, we identify CSF proteins that consistently predicted both short- and long-term disease progression. Lower levels of neurofilament light chain (NfL) in CSF is superior in predicting the absence of disease activity two years after sampling (replication AUC = 0.77) compared to all other tested proteins. Importantly, we also identify a combination of 11 CSF proteins (CXCL13, LTA, FCN2, ICAM3, LY9, SLAMF7, TYMP, CHI3L1, FYB1, TNFRSF1B and NfL) that predict the severity of disability worsening according to the normalized age-related MS severity score (replication AUC = 0.90). The identification of these proteins may help elucidate pathogenetic processes and might aid decisions on treatment strategies for persons with MS.
© 2023. The Author(s).
Conflict of interest statement
T.O. has received advisory board/lecture honoraria as well as unrestricted research grants from Biogen, Novartis, Sanofi, and Merck. None of which has any relation to the current manuscript. F.P. has received research grants from Janssen, Merck KgaA and UCB, and fees for serving on DMC in clinical trials with Chugai, Lundbeck and Roche, and preparation of expert witness statement for Novartis. J.M. has received honoraria for advisory boards for Sanofi Genzyme and Merck and lecture honorarium from Merck. The remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
