

Landscape of enhancer disruption and functional screen in melanoma cells
- PMID: 37904237
- PMCID: PMC10614365
- DOI: 10.1186/s13059-023-03087-5
Landscape of enhancer disruption and functional screen in melanoma cells
Abstract
Background: The high mutation rate throughout the entire melanoma genome presents a major challenge in stratifying true driver events from the background mutations. Numerous recurrent non-coding alterations, such as those in enhancers, can shape tumor evolution, thereby emphasizing the importance in systematically deciphering enhancer disruptions in melanoma.
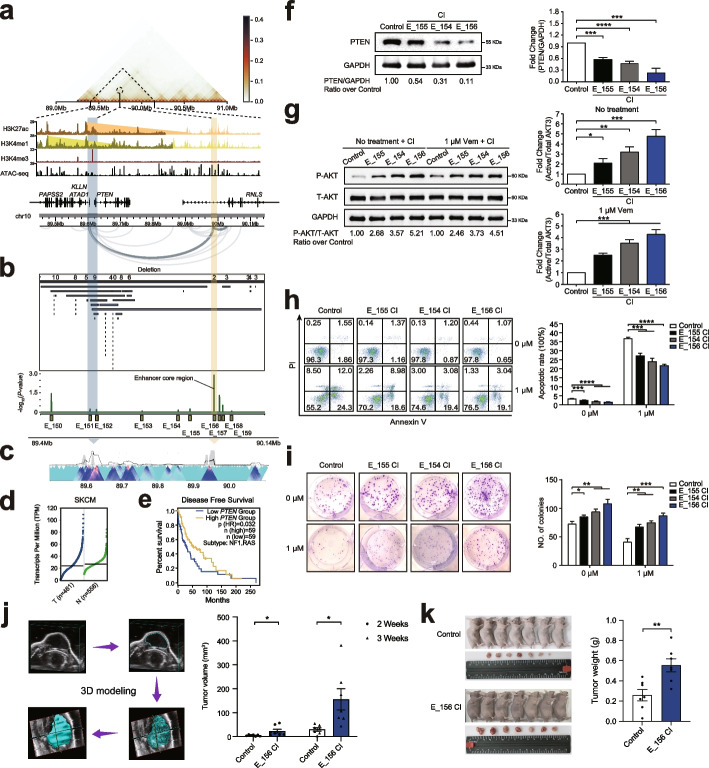
Results: Here, we leveraged 297 melanoma whole-genome sequencing samples to prioritize highly recurrent regions. By performing a genome-scale CRISPR interference (CRISPRi) screen on highly recurrent region-associated enhancers in melanoma cells, we identified 66 significant hits which could have tumor-suppressive roles. These functional enhancers show unique mutational patterns independent of classical significantly mutated genes in melanoma. Target gene analysis for the essential enhancers reveal many known and hidden mechanisms underlying melanoma growth. Utilizing extensive functional validation experiments, we demonstrate that a super enhancer element could modulate melanoma cell proliferation by targeting MEF2A, and another distal enhancer is able to sustain PTEN tumor-suppressive potential via long-range interactions.
Conclusions: Our study establishes a catalogue of crucial enhancers and their target genes in melanoma growth and progression, and illuminates the identification of novel mechanisms of dysregulation for melanoma driver genes and new therapeutic targeting strategies.
Keywords: Enhancer; Highly recurrent regions (HRRs); Melanoma; Myocyte enhancer factor 2A (MEF2A); Phosphatase and tensin homolog (PTEN).
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
