

This is a preprint.
Exploiting the Cullin E3 Ligase Adaptor Protein SKP1 for Targeted Protein Degradation
- PMID: 37904950
- PMCID: PMC10614948
- DOI: 10.1101/2023.10.20.563371
Exploiting the Cullin E3 Ligase Adaptor Protein SKP1 for Targeted Protein Degradation
Update in
-
Exploiting the Cullin E3 Ligase Adaptor Protein SKP1 for Targeted Protein Degradation.ACS Chem Biol. 2024 Feb 16;19(2):442-450. doi: 10.1021/acschembio.3c00642. Epub 2024 Feb 2. ACS Chem Biol. 2024. PMID: 38305738 Free PMC article.
Abstract
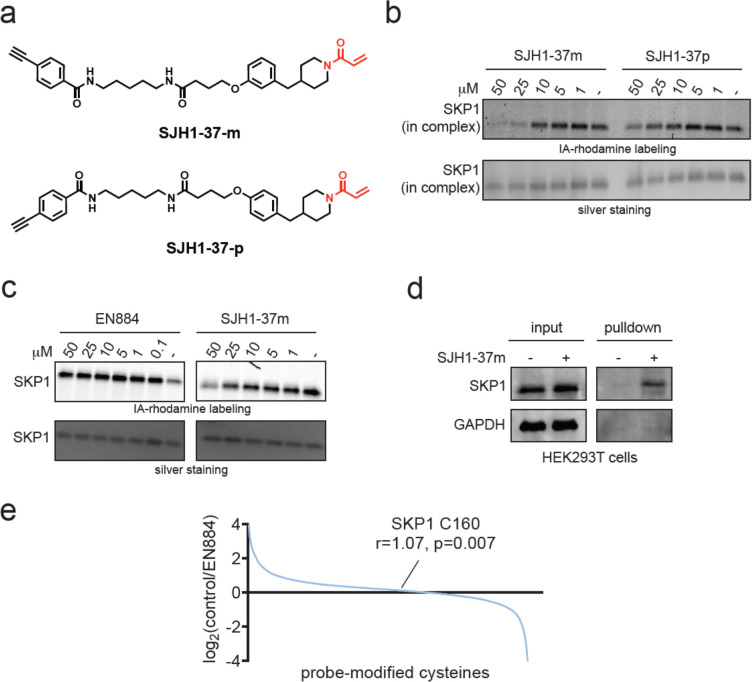
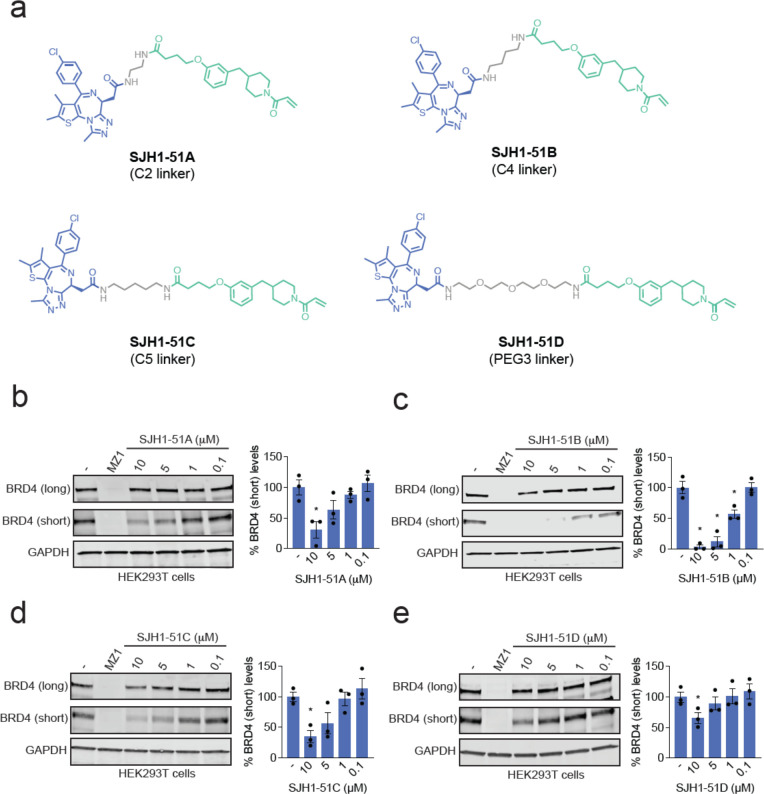
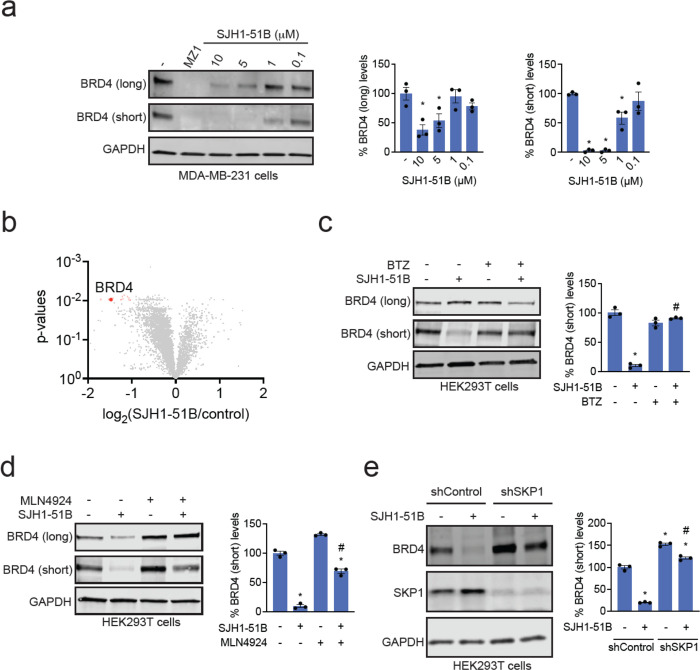
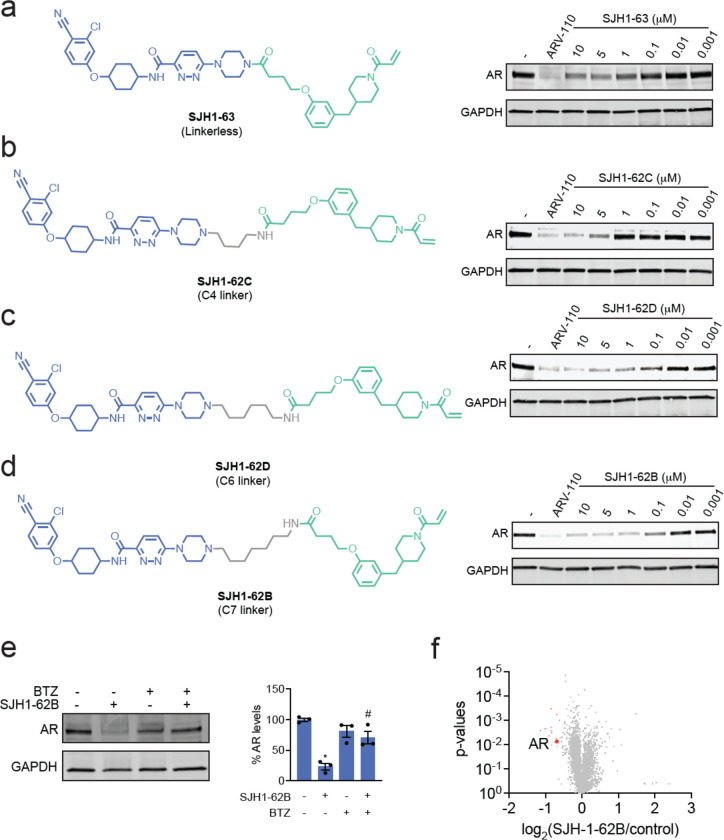
Targeted protein degradation with Proteolysis Targeting Chimeras (PROTACs) is a powerful therapeutic modality for eliminating disease-causing proteins through targeted ubiquitination and proteasome-mediated degradation. Most PROTACs have exploited substrate receptors of Cullin-RING E3 ubiquitin ligases such as cereblon and VHL. Whether core, shared, and essential components of the Cullin-RING E3 ubiquitin ligase complex can be used for PROTAC applications remains less explored. Here, we discovered a cysteine-reactive covalent recruiter EN884 against the SKP1 adapter protein of the SKP1-CUL1-F-box containing SCF complex. We further showed that this recruiter can be used in PROTAC applications to degrade neo-substrate proteins such as BRD4 and the androgen receptor in a SKP1- and proteasome-dependent manner. Our studies demonstrate that core and essential adapter proteins within the Cullin-RING E3 ubiquitin ligase complex can be exploited for targeted protein degradation applications and that covalent chemoproteomic strategies can enable recruiter discovery against these targets.
Keywords: AR; BRD4; CUL1; SKP1; activity-based protein profiling; androgen receptor; chemoproteomics; covalent; targeted protein degradation.
Conflict of interest statement
Competing Financial Interests Statement OWH was an employee of Bristol Myers Squibb when this study was initiated, but is now an employee of Lyterian Therapeutics. IEW was an employee of Bristol Myers Squibb when this study was initiated but is now a co-founder and the CEO of Lyterian Therapeutics. IEW is on the Scientific Advisory Boards of PAIVBio and Firefly Biologics. DKN is a co-founder, shareholder, and scientific advisory board member for Frontier Medicines and Vicinitas Therapeutics. DKN is a member of the board of directors for Vicinitas Therapeutics. DKN is also on the scientific advisory board of The Mark Foundation for Cancer Research, Photys Therapeutics, and Apertor Pharmaceuticals. DKN is also an Investment Advisory Partner for a16z Bio, an Advisory Board member for Droia Ventures, and an iPartner for The Column Group.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials