

This is a preprint.
Pan-cancer analysis reveals multifaceted roles of retrotransposon-fusion RNAs
- PMID: 37905014
- PMCID: PMC10614793
- DOI: 10.1101/2023.10.16.562422
Pan-cancer analysis reveals multifaceted roles of retrotransposon-fusion RNAs
Abstract
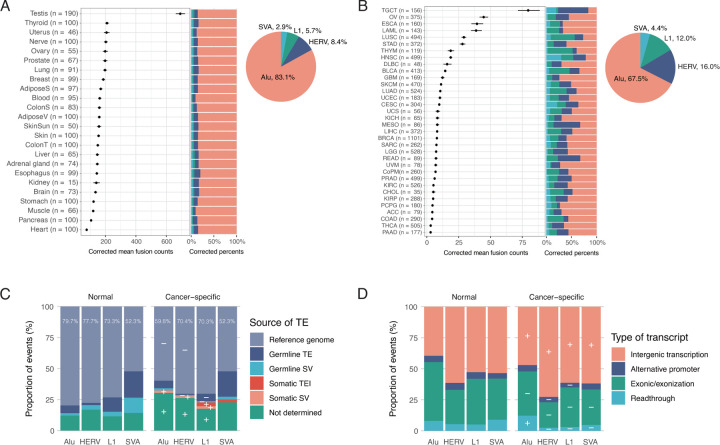
Transposon-derived transcripts are abundant in RNA sequences, yet their landscape and function, especially for fusion transcripts derived from unannotated or somatically acquired transposons, remains underexplored. Here, we developed a new bioinformatic tool to detect transposon-fusion transcripts in RNA-sequencing data and performed a pan-cancer analysis of 10,257 cancer samples across 34 cancer types as well as 3,088 normal tissue samples. We identified 52,277 cancer-specific fusions with ~30 events per cancer and hotspot loci within transposons vulnerable to fusion formation. Exonization of intronic transposons was the most prevalent genic fusions, while somatic L1 insertions constituted a small fraction of cancer-specific fusions. Source L1s and HERVs, but not Alus showed decreased DNA methylation in cancer upon fusion formation. Overall cancer-specific L1 fusions were enriched in tumor suppressors while Alu fusions were enriched in oncogenes, including recurrent Alu fusions in EZH2 predictive of patient survival. We also demonstrated that transposon-derived peptides triggered CD8+ T-cell activation to the extent comparable to EBV viruses. Our findings reveal distinct epigenetic and tumorigenic mechanisms underlying transposon fusions across different families and highlight transposons as novel therapeutic targets and the source of potent neoantigens.
Keywords: Cancer neoantigen; Gene fusion; Integrative multiomics data; Pan-cancer analysis; Transposable element.
Conflict of interest statement
Competing interests W-Y.P. is a founder and CEO of Geninus Inc. The other authors declare no competing interests.
Figures




References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials