

Single-cell transcriptomics and chromatin accessibility profiling elucidate the kidney-protective mechanism of mineralocorticoid receptor antagonists
- PMID: 37906287
- PMCID: PMC10760974
- DOI: 10.1172/JCI157165
Single-cell transcriptomics and chromatin accessibility profiling elucidate the kidney-protective mechanism of mineralocorticoid receptor antagonists
Abstract
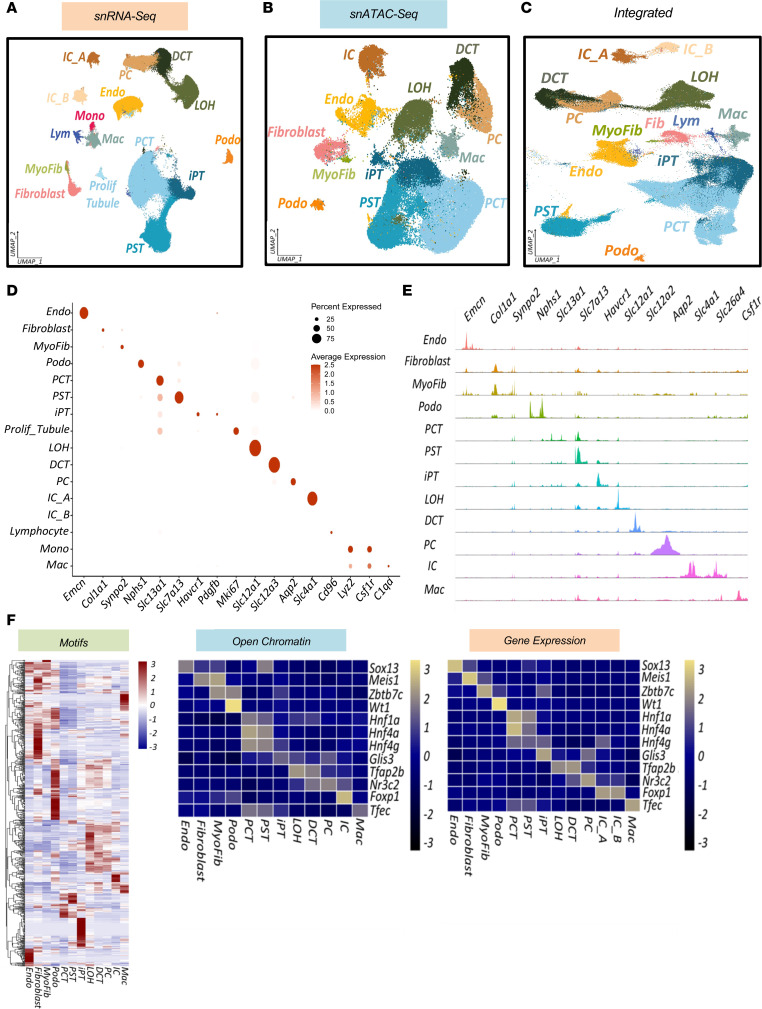
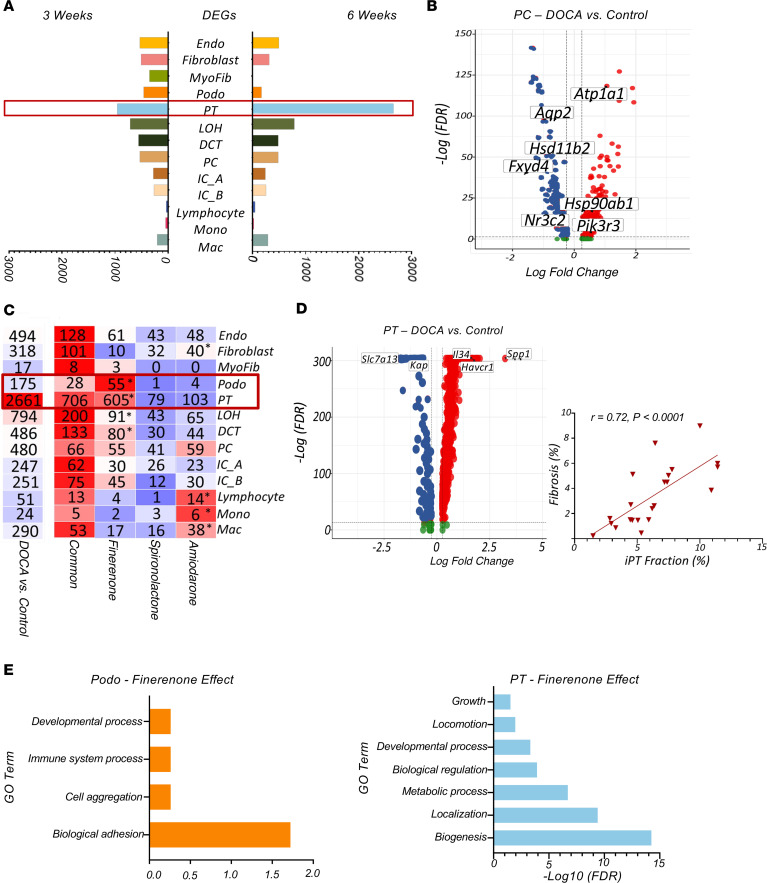
Mineralocorticoid excess commonly leads to hypertension (HTN) and kidney disease. In our study, we used single-cell expression and chromatin accessibility tools to characterize the mineralocorticoid target genes and cell types. We demonstrated that mineralocorticoid effects were established through open chromatin and target gene expression, primarily in principal and connecting tubule cells and, to a lesser extent, in segments of the distal convoluted tubule cells. We examined the kidney-protective effects of steroidal and nonsteroidal mineralocorticoid antagonists (MRAs), as well as of amiloride, an epithelial sodium channel inhibitor, in a rat model of deoxycorticosterone acetate, unilateral nephrectomy, and high-salt consumption-induced HTN and cardiorenal damage. All antihypertensive therapies protected against cardiorenal damage. However, finerenone was particularly effective in reducing albuminuria and improving gene expression changes in podocytes and proximal tubule cells, even with an equivalent reduction in blood pressure. We noted a strong correlation between the accumulation of injured/profibrotic tubule cells expressing secreted posphoprotein 1 (Spp1), Il34, and platelet-derived growth factor subunit b (Pdgfb) and the degree of fibrosis in rat kidneys. This gene signature also showed a potential for classifying human kidney samples. Our multiomics approach provides fresh insights into the possible mechanisms underlying HTN-associated kidney disease, the target cell types, the protective effects of steroidal and nonsteroidal MRAs, and amiloride.
Keywords: Chronic kidney disease; Genetics; Molecular genetics; Nephrology.
Conflict of interest statement
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
