

Metabolomic Rewiring Promotes Endocrine Therapy Resistance in Breast Cancer
- PMID: 37906431
- PMCID: PMC10842725
- DOI: 10.1158/0008-5472.CAN-23-0184
Metabolomic Rewiring Promotes Endocrine Therapy Resistance in Breast Cancer
Abstract
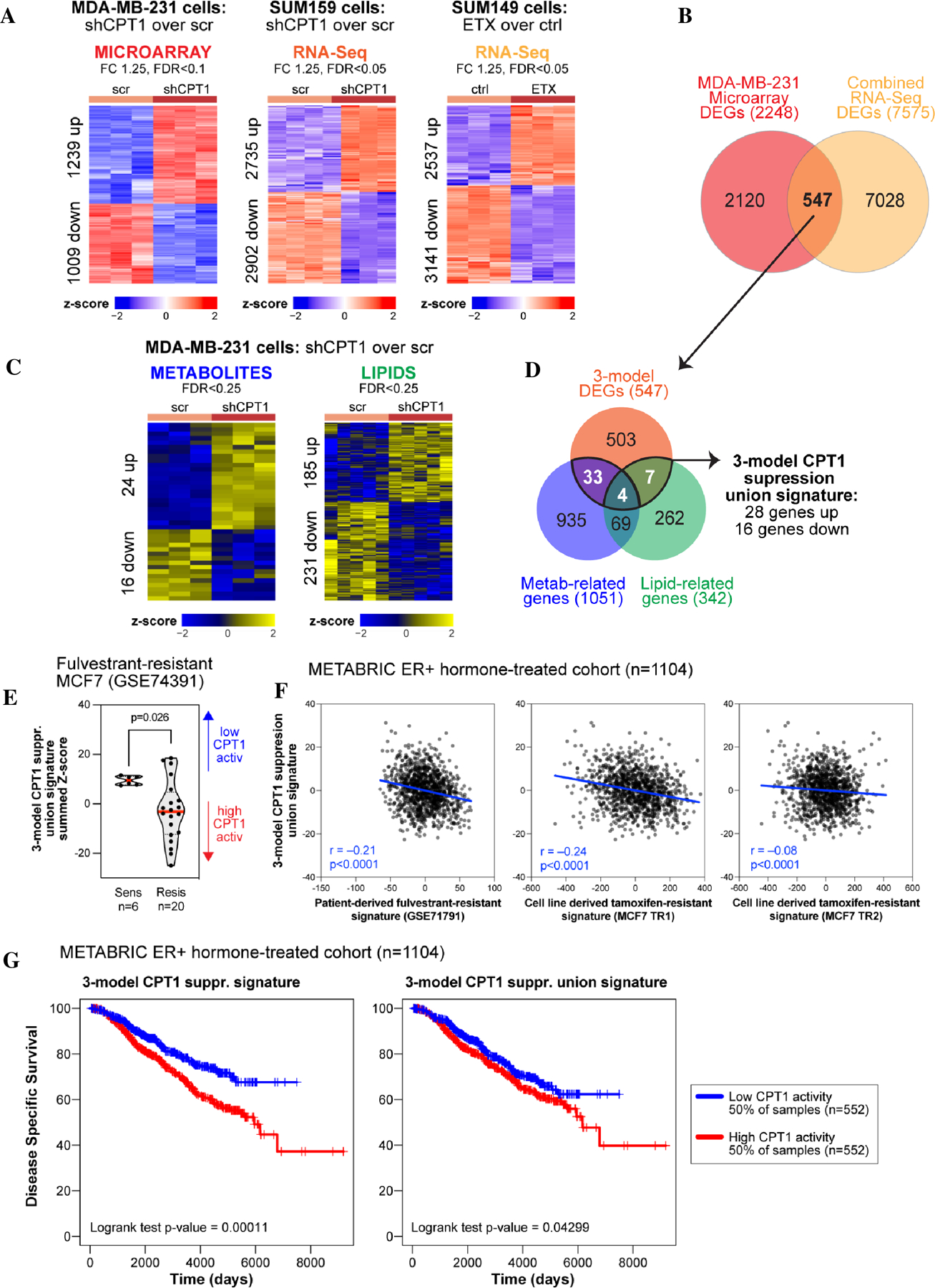
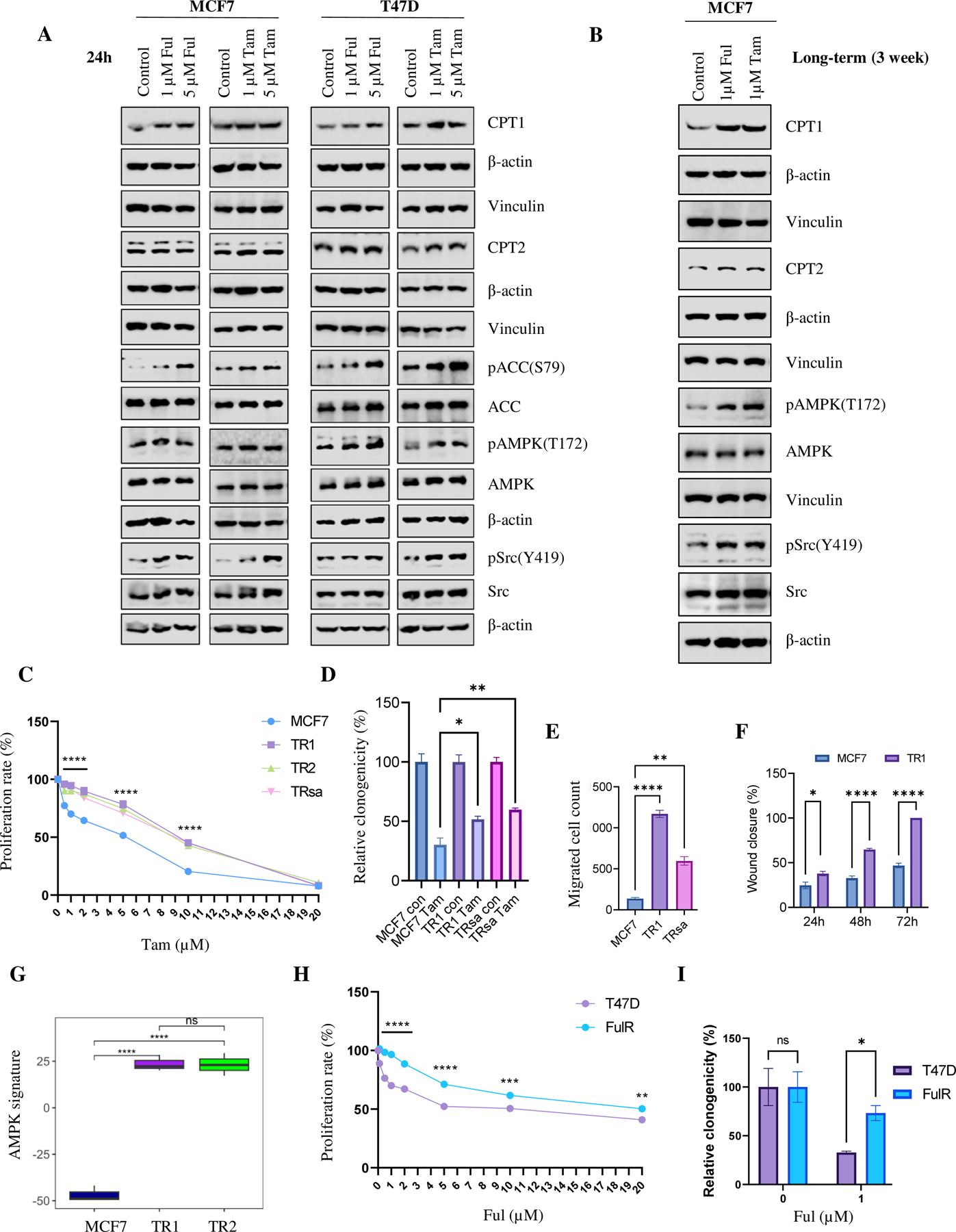
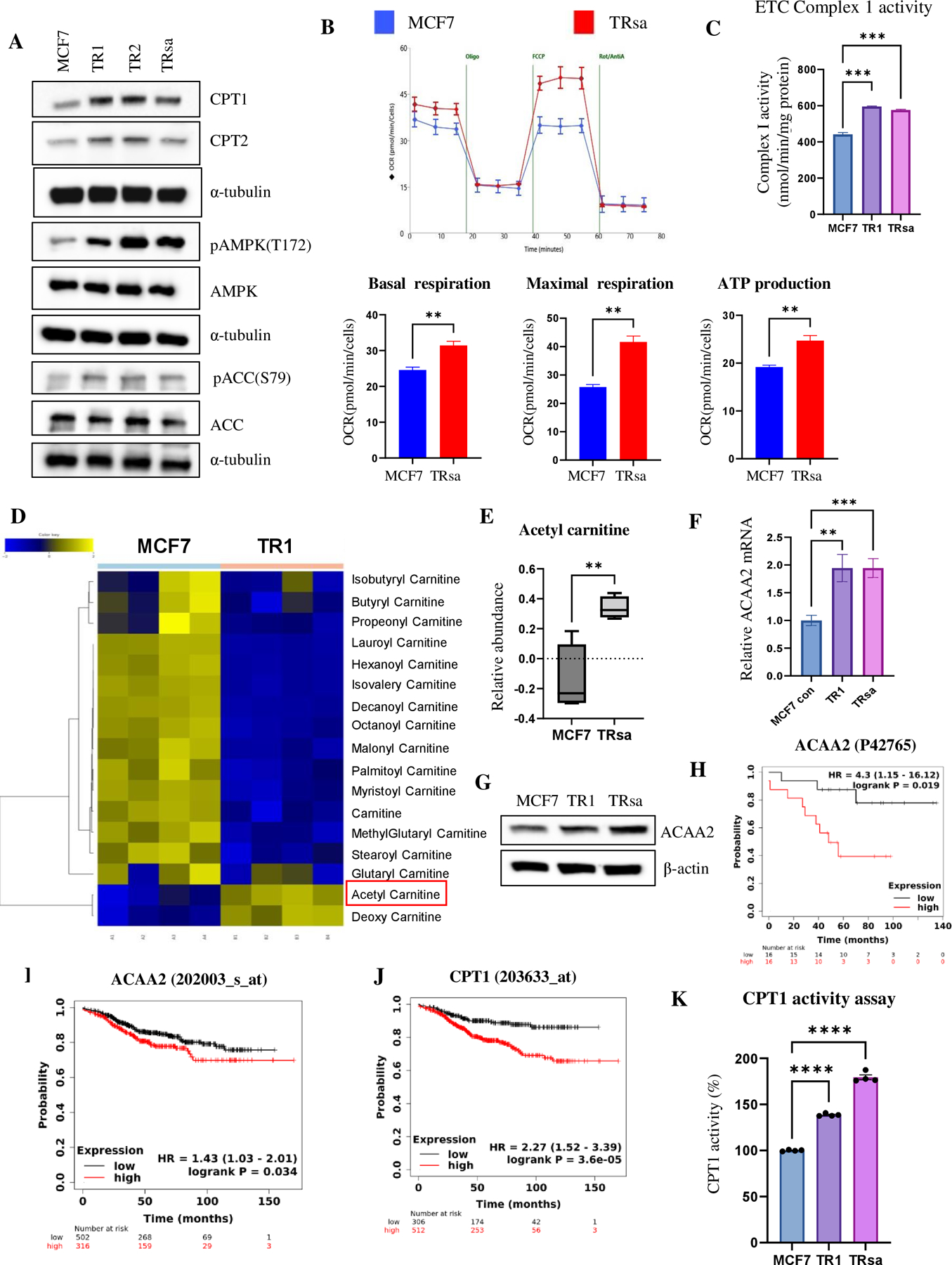
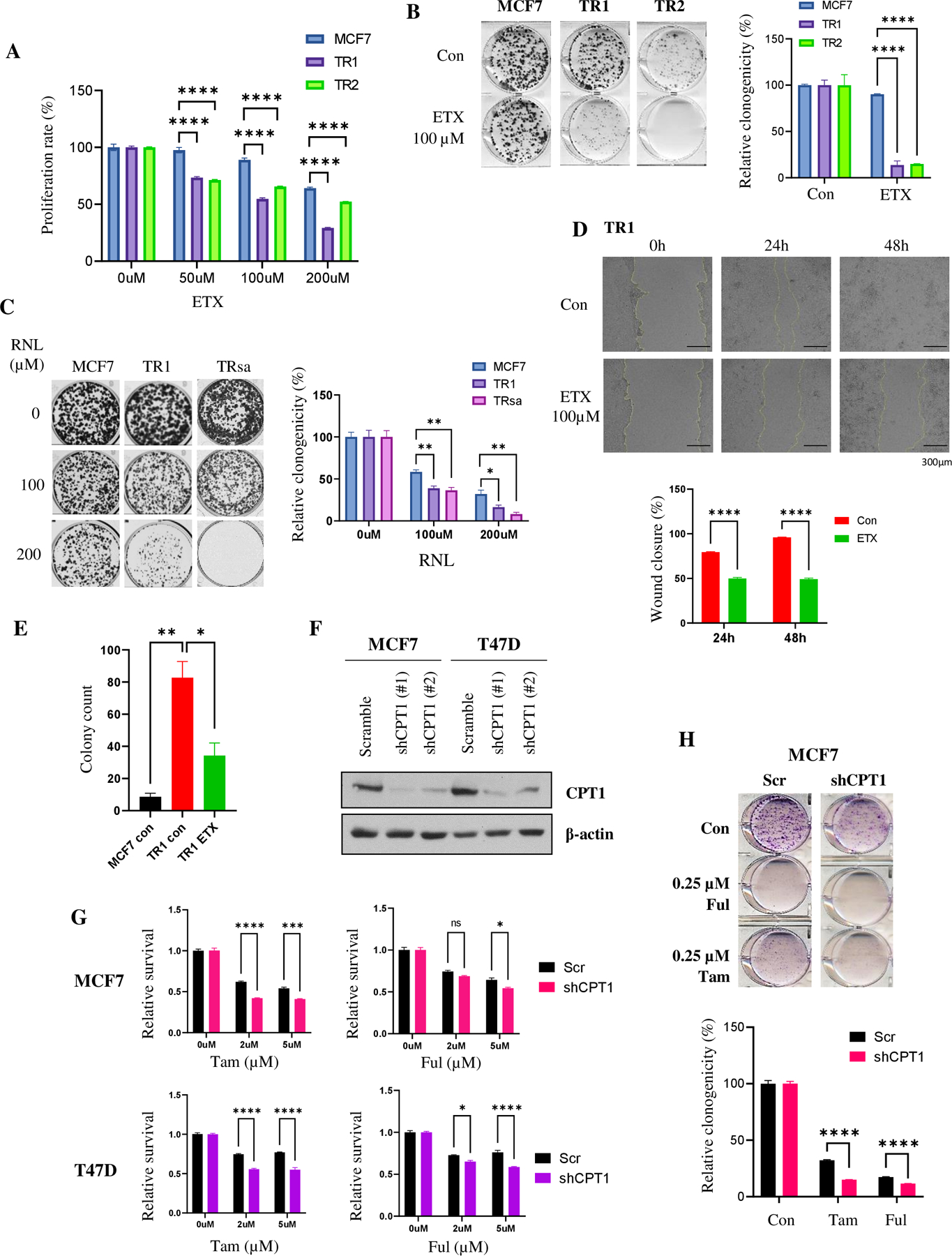
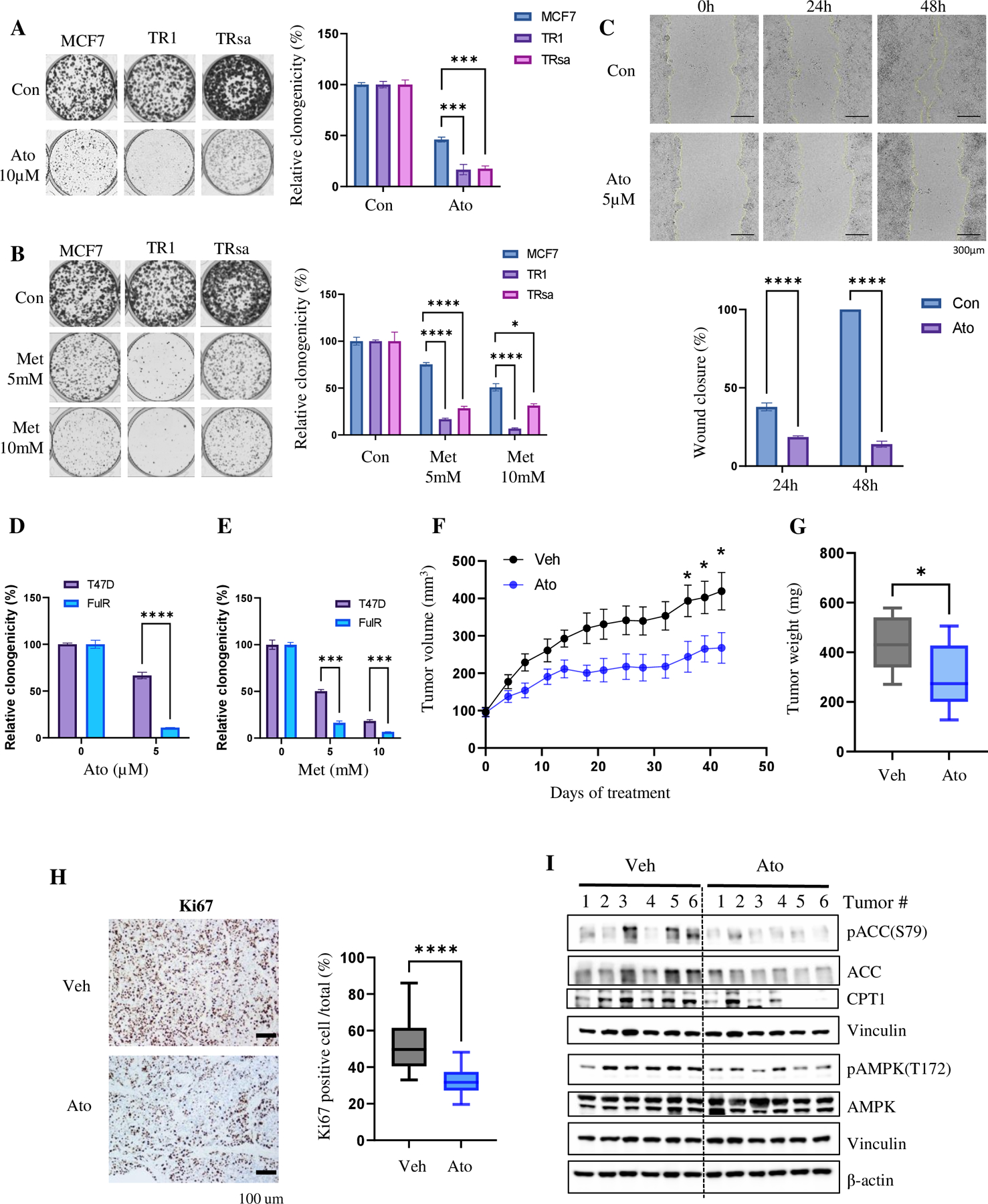
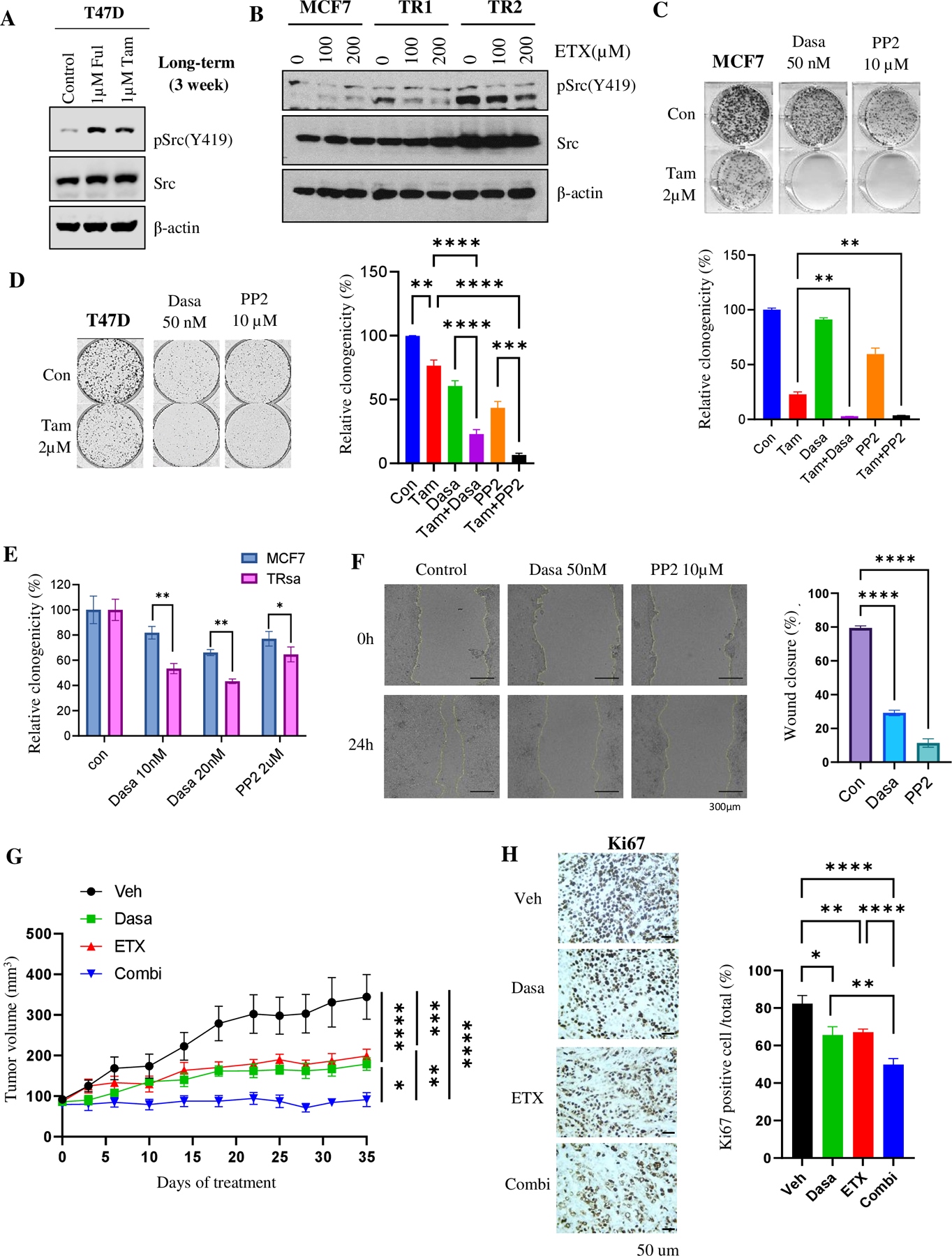
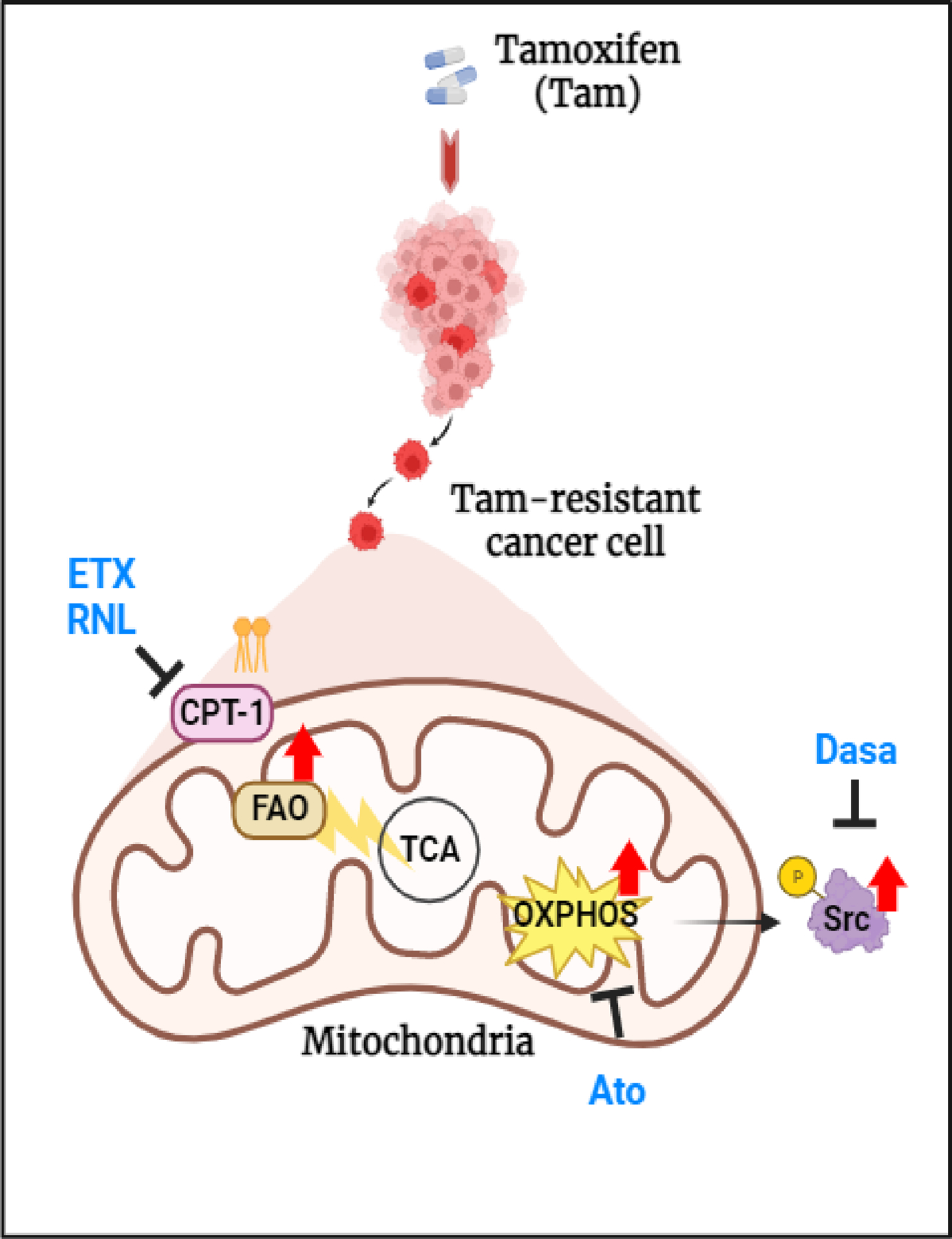
Approximately one-third of endocrine-treated women with estrogen receptor alpha-positive (ER+) breast cancers are at risk of recurrence due to intrinsic or acquired resistance. Thus, it is vital to understand the mechanisms underlying endocrine therapy resistance in ER+ breast cancer to improve patient treatment. Mitochondrial fatty acid β-oxidation (FAO) has been shown to be a major metabolic pathway in triple-negative breast cancer (TNBC) that can activate Src signaling. Here, we found metabolic reprogramming that increases FAO in ER+ breast cancer as a mechanism of resistance to endocrine therapy. A metabolically relevant, integrated gene signature was derived from transcriptomic, metabolomic, and lipidomic analyses in TNBC cells following inhibition of the FAO rate-limiting enzyme carnitine palmitoyl transferase 1 (CPT1), and this TNBC-derived signature was significantly associated with endocrine resistance in patients with ER+ breast cancer. Molecular, genetic, and metabolomic experiments identified activation of AMPK-FAO-oxidative phosphorylation (OXPHOS) signaling in endocrine-resistant ER+ breast cancer. CPT1 knockdown or treatment with FAO inhibitors in vitro and in vivo significantly enhanced the response of ER+ breast cancer cells to endocrine therapy. Consistent with the previous findings in TNBC, endocrine therapy-induced FAO activated the Src pathway in ER+ breast cancer. Src inhibitors suppressed the growth of endocrine-resistant tumors, and the efficacy could be further enhanced by metabolic priming with CPT1 inhibition. Collectively, this study developed and applied a TNBC-derived signature to reveal that metabolic reprogramming to FAO activates the Src pathway to drive endocrine resistance in ER+ breast cancer.
Significance: Increased fatty acid oxidation induced by endocrine therapy activates Src signaling to promote endocrine resistance in breast cancer, which can be overcome using clinically approved therapies targeting FAO and Src.
©2023 American Association for Cancer Research.
Conflict of interest statement
Conflicts of interest
The authors of this manuscript have no conflict of interest to declare
Figures
References
-
- Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003;22:7316–39 - PubMed
-
- Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev 2004;25:869–98 - PubMed
-
- deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res 2004;10:8059–67 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
