

Toxic PARP trapping upon cAMP-induced DNA damage reinstates the efficacy of endocrine therapy and CDK4/6 inhibitors in treatment-refractory ER+ breast cancer
- PMID: 37914699
- PMCID: PMC10620179
- DOI: 10.1038/s41467-023-42736-y
Toxic PARP trapping upon cAMP-induced DNA damage reinstates the efficacy of endocrine therapy and CDK4/6 inhibitors in treatment-refractory ER+ breast cancer
Abstract
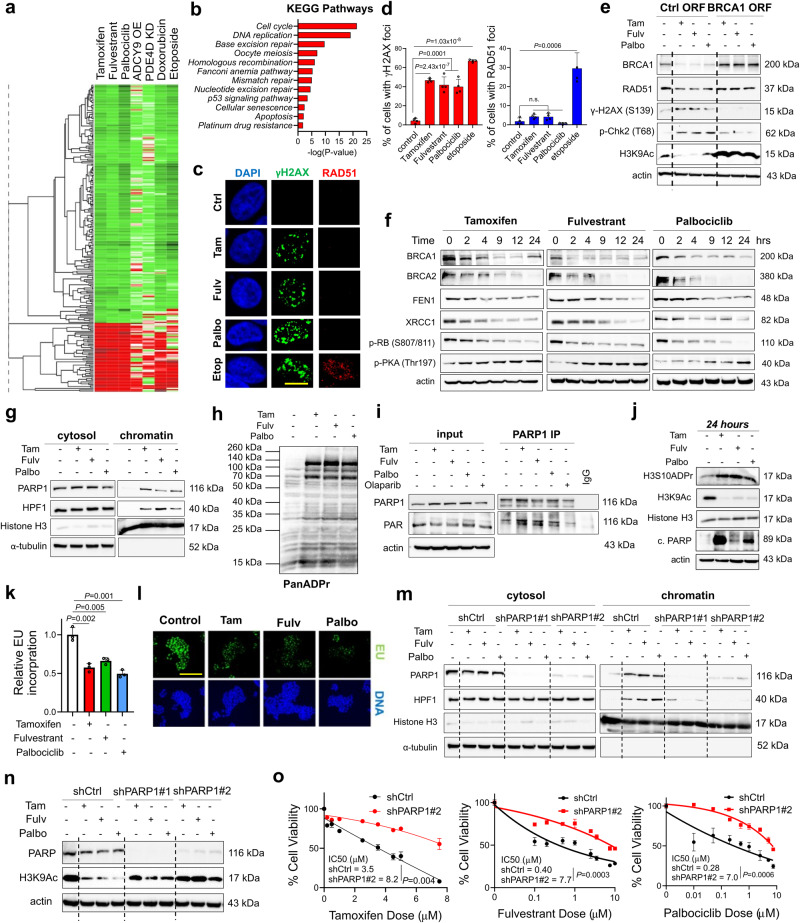
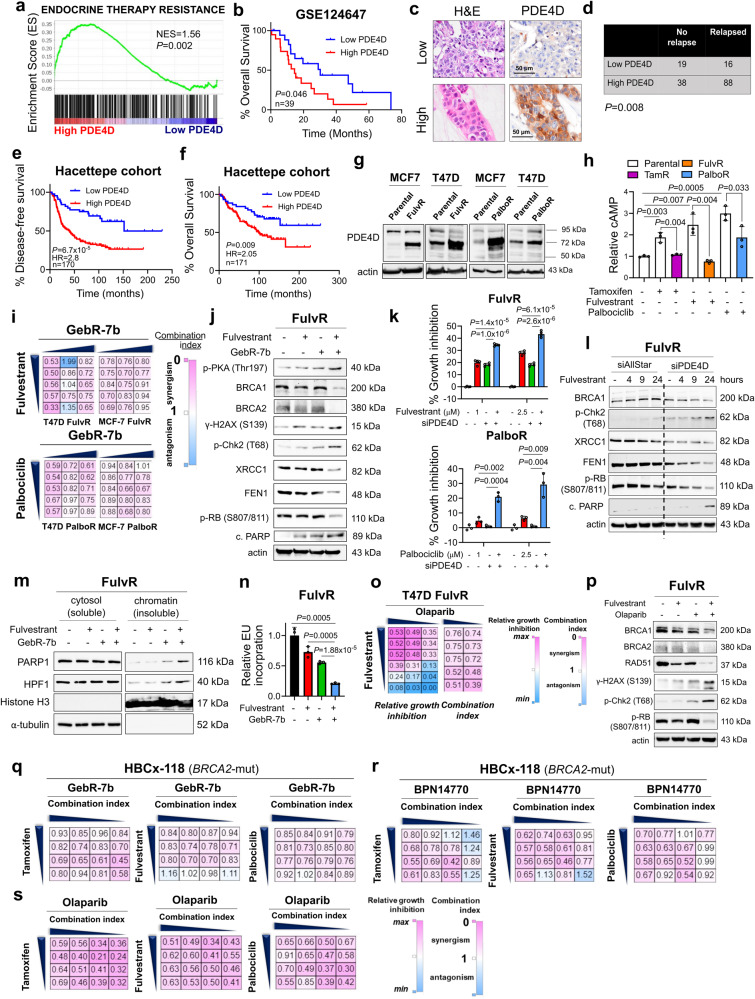
Resistance to endocrine therapy and CDK4/6 inhibitors, the standard of care (SOC) in estrogen receptor-positive (ER+) breast cancer, greatly reduces patient survival. Therefore, elucidating the mechanisms of sensitivity and resistance to SOC therapy and identifying actionable targets are urgently needed. Here, we show that SOC therapy causes DNA damage and toxic PARP1 trapping upon generation of a functional BRCAness (i.e., BRCA1/2 deficiency) phenotype, leading to increased histone parylation and reduced H3K9 acetylation, resulting in transcriptional blockage and cell death. Mechanistically, SOC therapy downregulates phosphodiesterase 4D (PDE4D), a novel ER target gene in a feedforward loop with ER, resulting in increased cAMP, PKA-dependent phosphorylation of mitochondrial COXIV-I, ROS generation and DNA damage. However, during SOC resistance, an ER-to-EGFR switch induces PDE4D overexpression via c-Jun. Notably, combining SOC with inhibitors of PDE4D, EGFR or PARP1 overcomes SOC resistance irrespective of the BRCA1/2 status, providing actionable targets for restoring SOC efficacy.
© 2023. The Author(s).
Conflict of interest statement
O. Sahin is the co-founder and manager of OncoCube Therapeutics LLC, developing TACC3 inhibitors, and the founder and president of LoxiGen, Inc, developing lysyl oxidase inhibitors. The other authors declare no potential conflicts of interest.
Figures







References
-
- Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. - PubMed
-
- DeSantis CE, et al. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019;69:438–451. - PubMed
-
- Lin NU, Winer EP. Advances in adjuvant endocrine therapy for postmenopausal women. J. Clin. Oncol. 2008;26:798–805. - PubMed
-
- Lloyd MR, Spring LM, Bardia A, Wander SA. Mechanisms of Resistance to CDK4/6 Blockade in Advanced Hormone Receptor-positive, HER2-negative Breast Cancer and Emerging Therapeutic Opportunities. Clin. Cancer Res. 2022;28:821–830. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
