

Glioma synapses recruit mechanisms of adaptive plasticity
- PMID: 37914930
- PMCID: PMC10632140
- DOI: 10.1038/s41586-023-06678-1
Glioma synapses recruit mechanisms of adaptive plasticity
Abstract
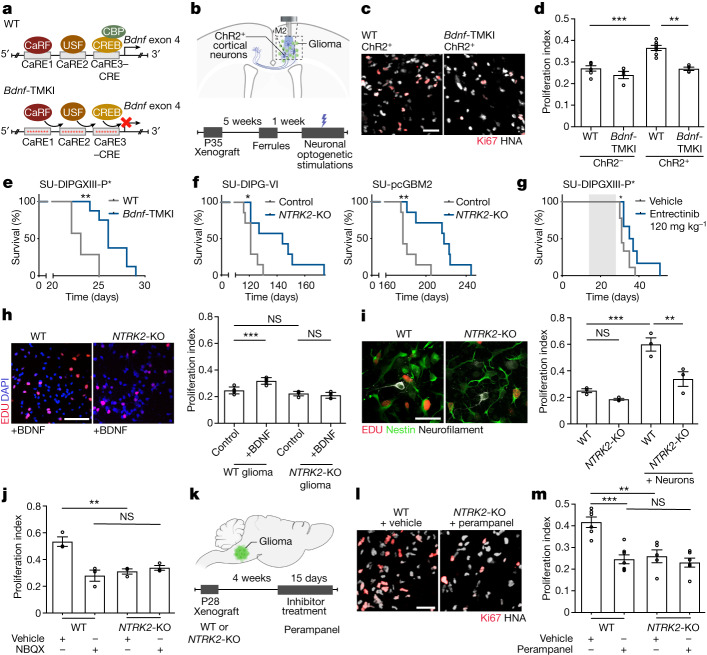
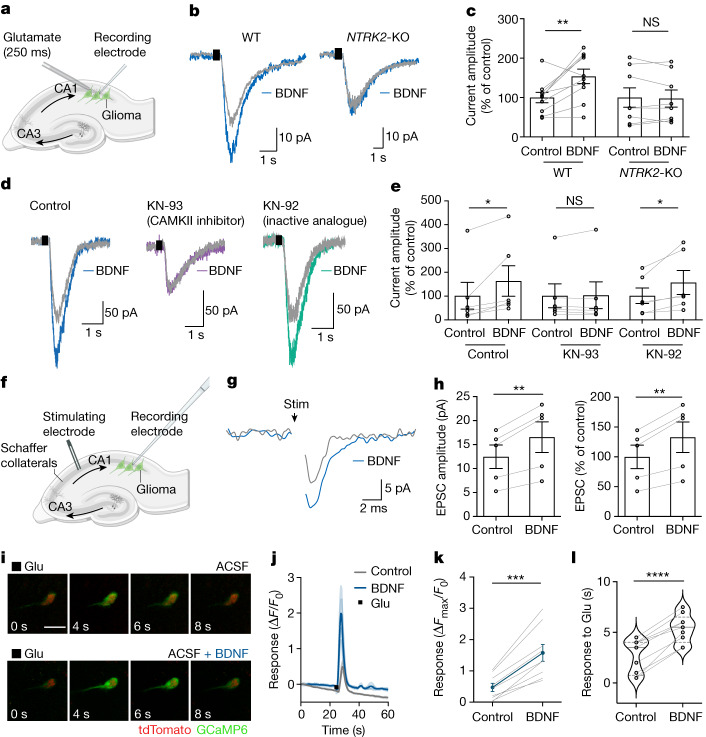
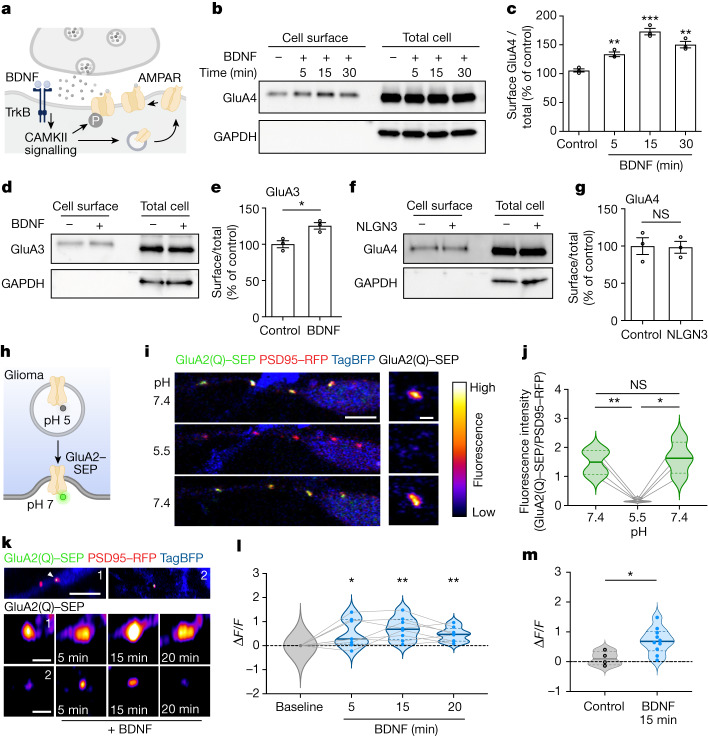
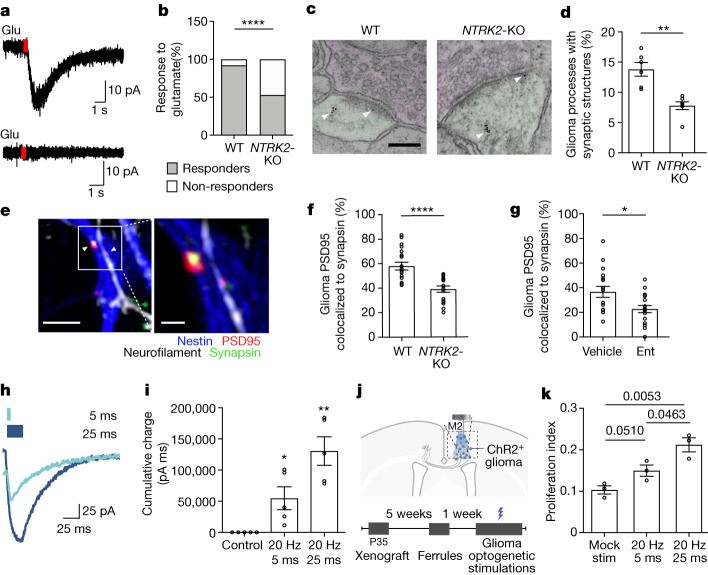
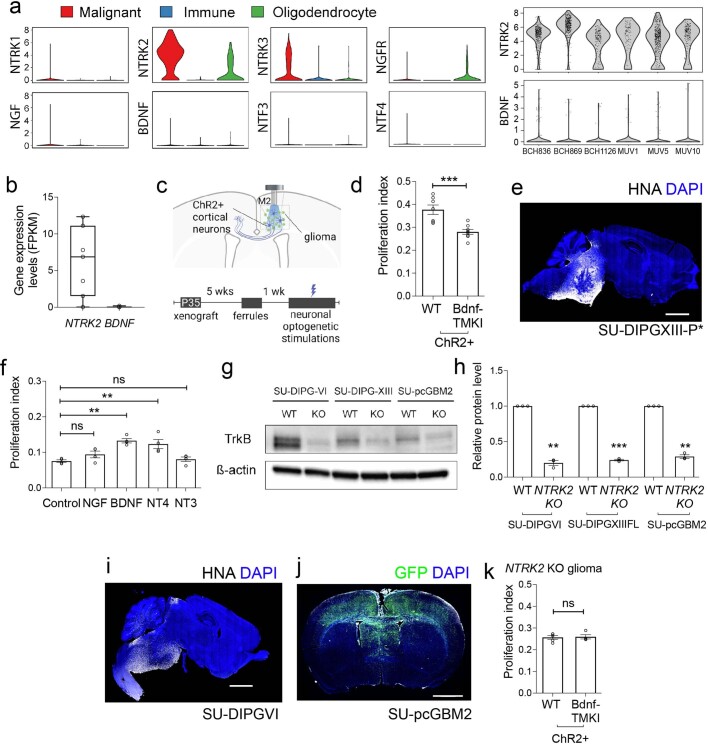
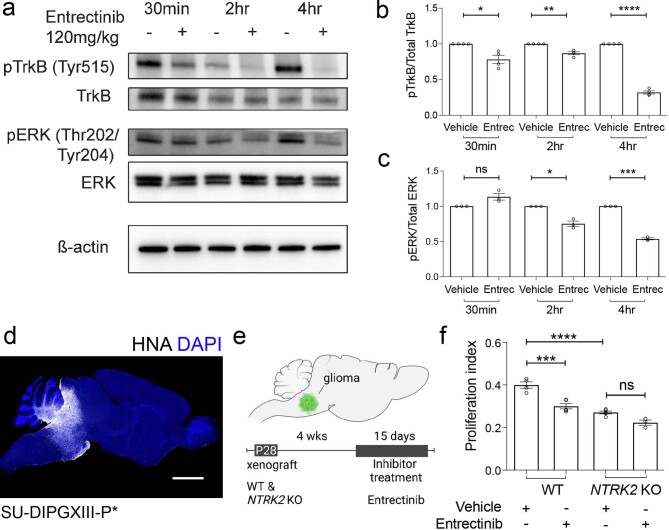
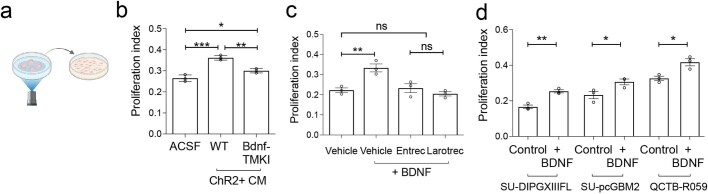
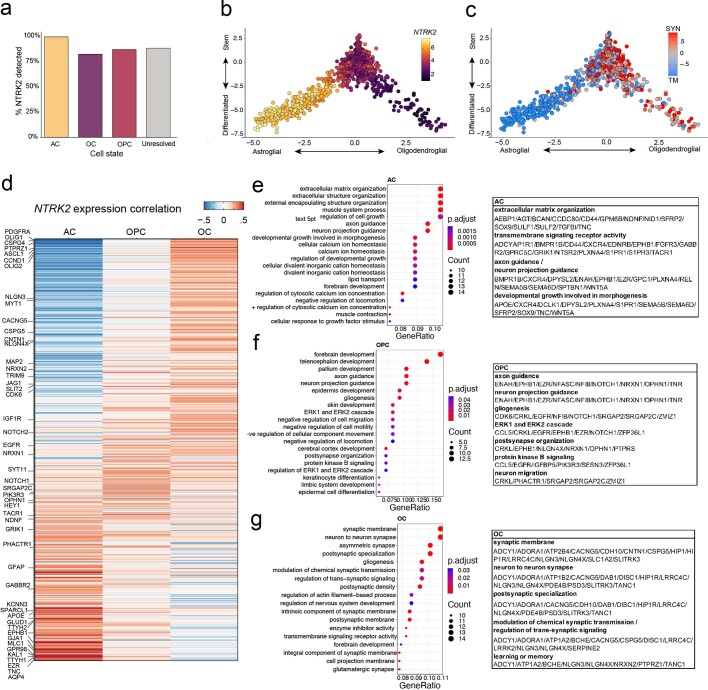
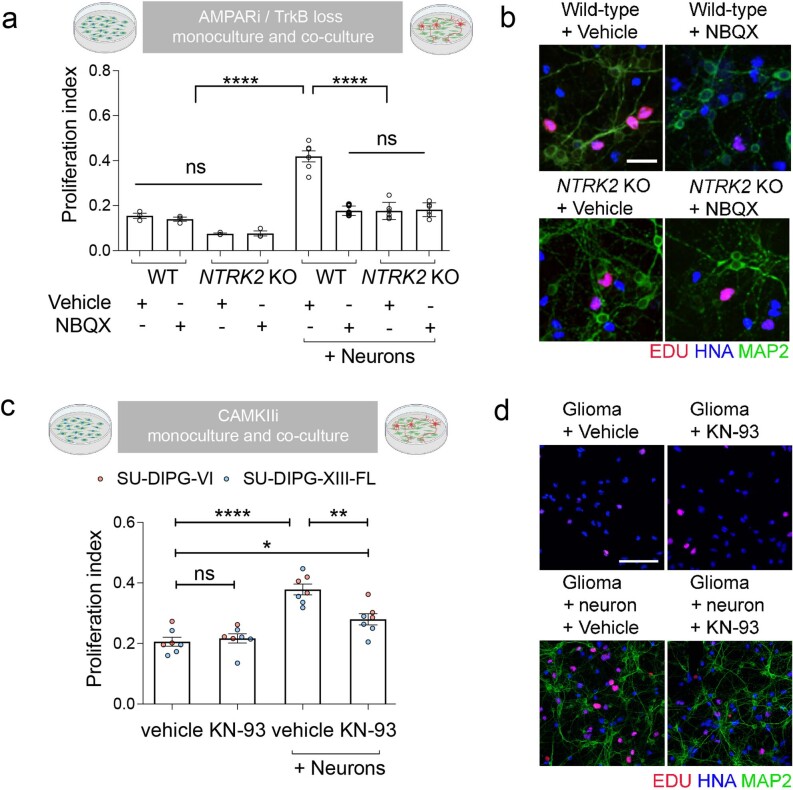
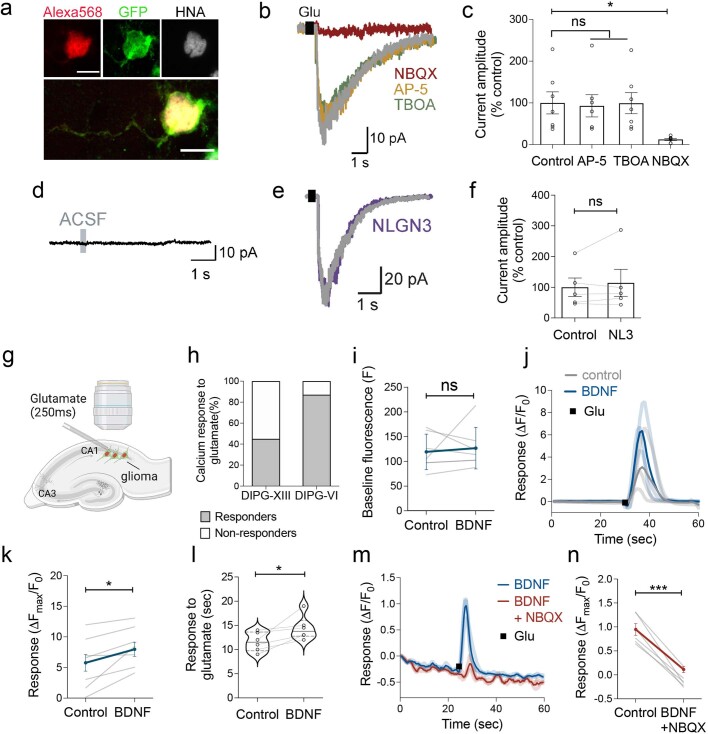
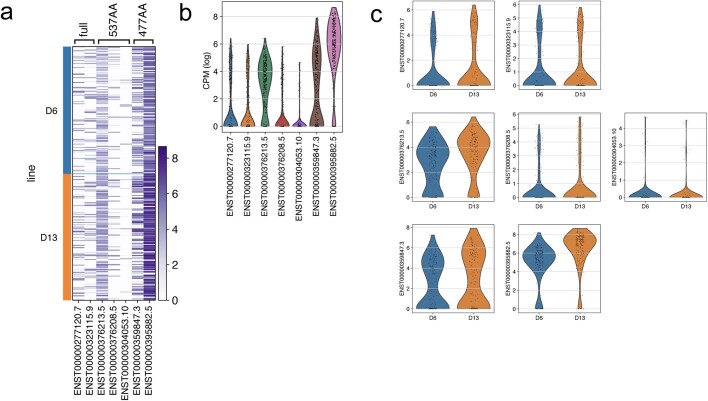
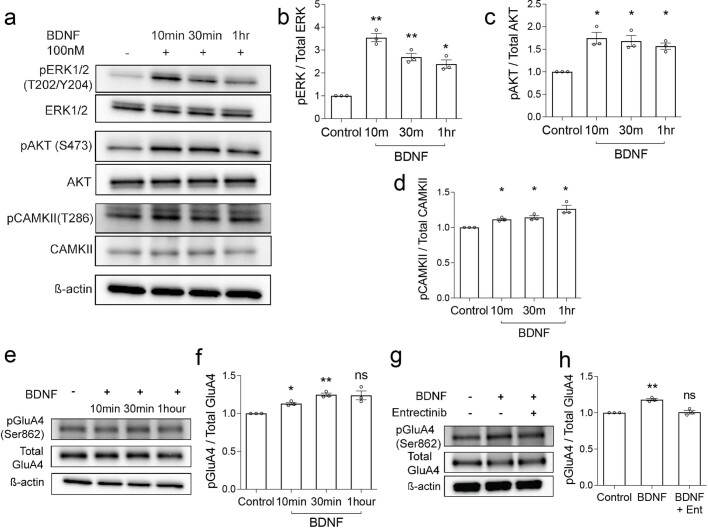
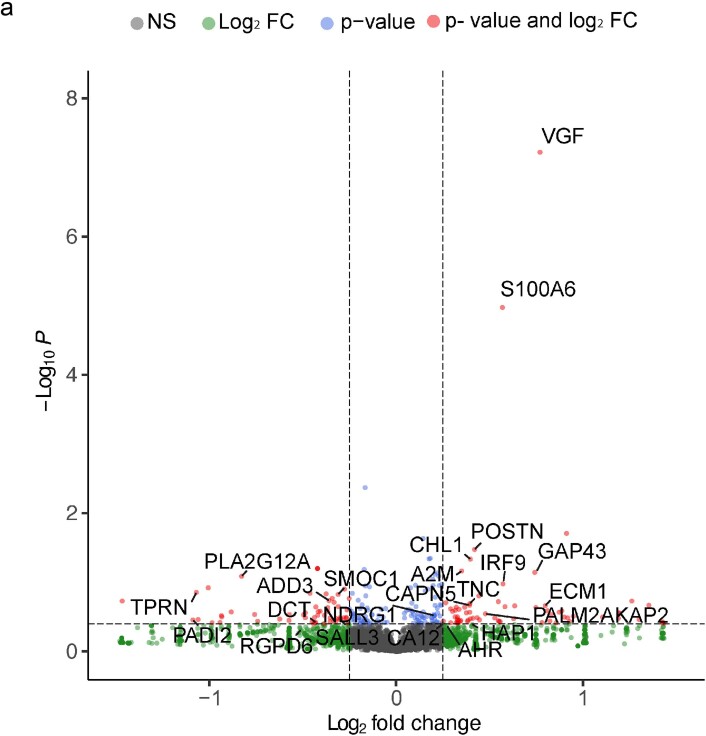
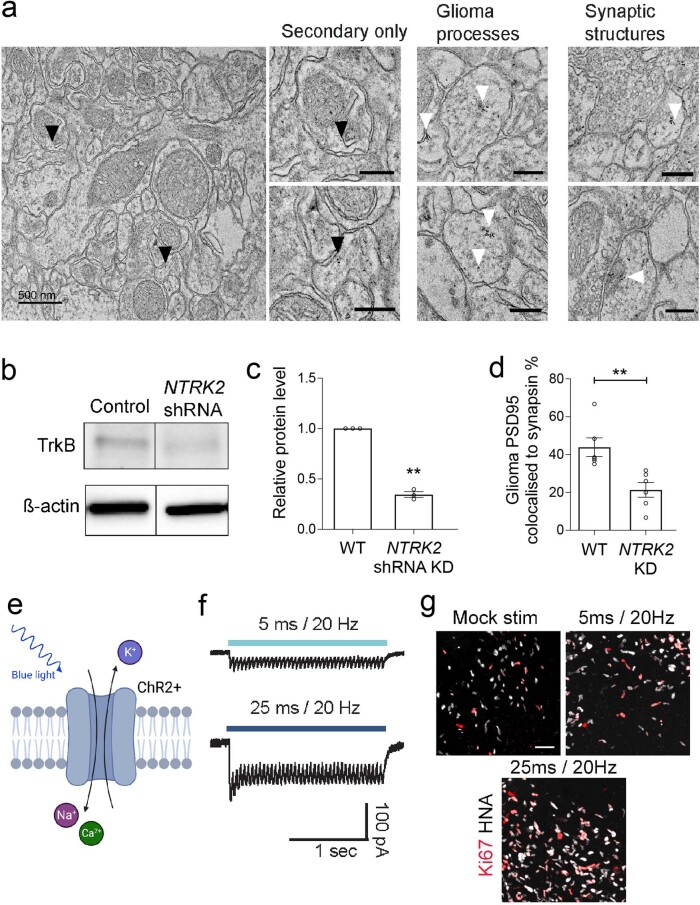
The role of the nervous system in the regulation of cancer is increasingly appreciated. In gliomas, neuronal activity drives tumour progression through paracrine signalling factors such as neuroligin-3 and brain-derived neurotrophic factor1-3 (BDNF), and also through electrophysiologically functional neuron-to-glioma synapses mediated by AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors4,5. The consequent glioma cell membrane depolarization drives tumour proliferation4,6. In the healthy brain, activity-regulated secretion of BDNF promotes adaptive plasticity of synaptic connectivity7,8 and strength9-15. Here we show that malignant synapses exhibit similar plasticity regulated by BDNF. Signalling through the receptor tropomyosin-related kinase B16 (TrkB) to CAMKII, BDNF promotes AMPA receptor trafficking to the glioma cell membrane, resulting in increased amplitude of glutamate-evoked currents in the malignant cells. Linking plasticity of glioma synaptic strength to tumour growth, graded optogenetic control of glioma membrane potential demonstrates that greater depolarizing current amplitude promotes increased glioma proliferation. This potentiation of malignant synaptic strength shares mechanistic features with synaptic plasticity17-22 that contributes to memory and learning in the healthy brain23-26. BDNF-TrkB signalling also regulates the number of neuron-to-glioma synapses. Abrogation of activity-regulated BDNF secretion from the brain microenvironment or loss of glioma TrkB expression robustly inhibits tumour progression. Blocking TrkB genetically or pharmacologically abrogates these effects of BDNF on glioma synapses and substantially prolongs survival in xenograft models of paediatric glioblastoma and diffuse intrinsic pontine glioma. Together, these findings indicate that BDNF-TrkB signalling promotes malignant synaptic plasticity and augments tumour progression.
© 2023. The Author(s).
Conflict of interest statement
M.M. was on the scientific advisory board for Cygnal Therapeutics, is on the scientific advisory board for TippingPoint Biosciences, and holds equity in MapLight Therapeutics. M.L.S. is an equity holder, scientific co-founder and advisory board member of Immunitas Therapeutics. The other authors declare no competing interests.
Figures
Comment in
-
Brain cancer thrives by hijacking mechanisms to boost synapse strength.Nature. 2023 Nov;623(7986):260-262. doi: 10.1038/d41586-023-03306-w. Nature. 2023. PMID: 37932559 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
