

Epigenetic regulation during cancer transitions across 11 tumour types
- PMID: 37914932
- PMCID: PMC10632147
- DOI: 10.1038/s41586-023-06682-5
Epigenetic regulation during cancer transitions across 11 tumour types
Abstract
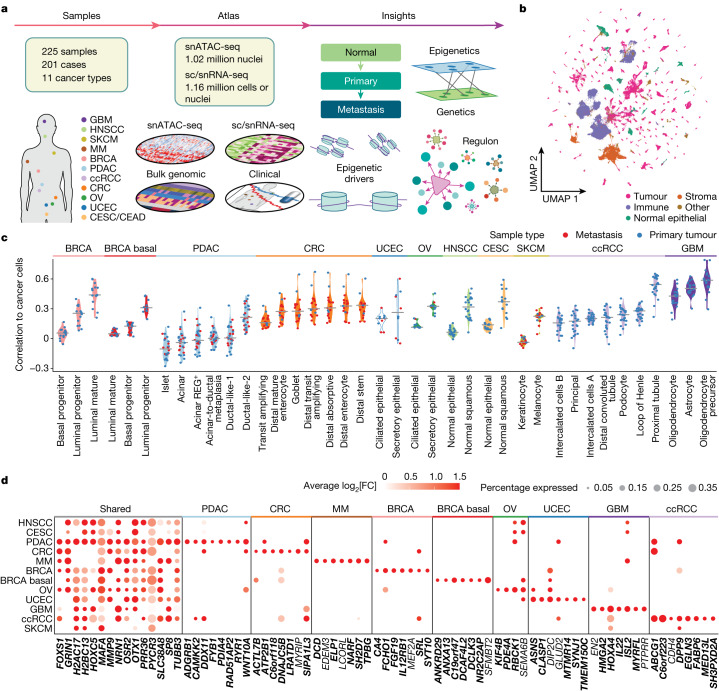
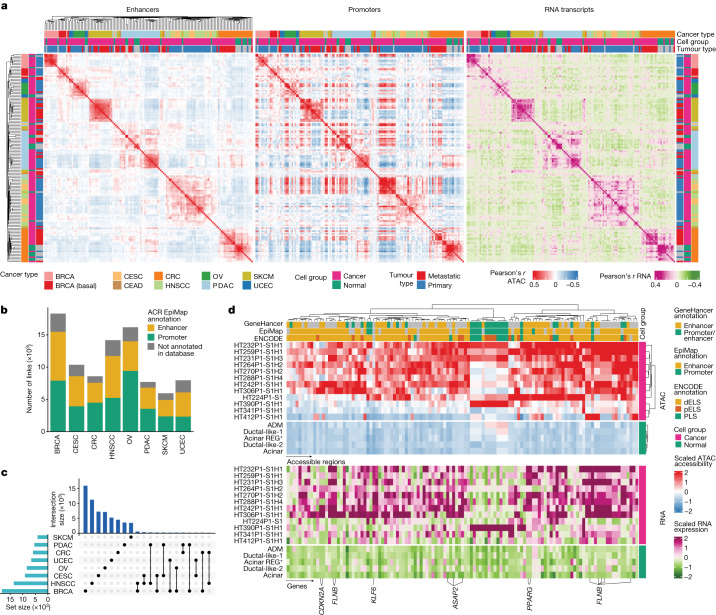
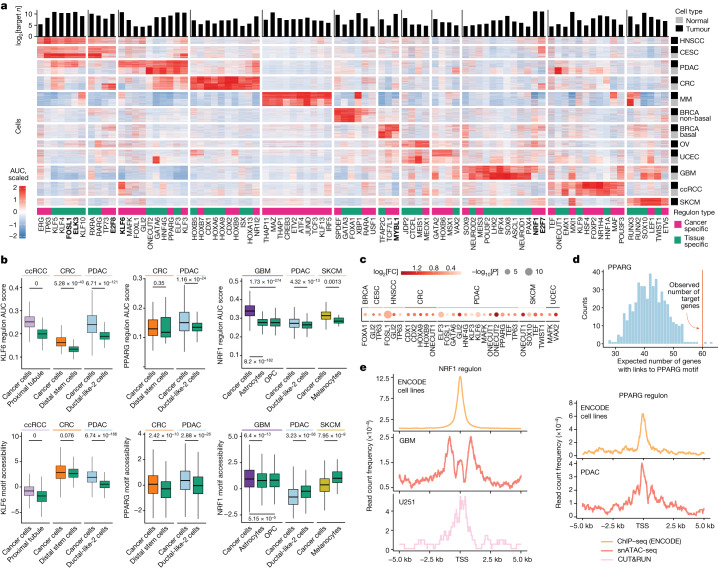
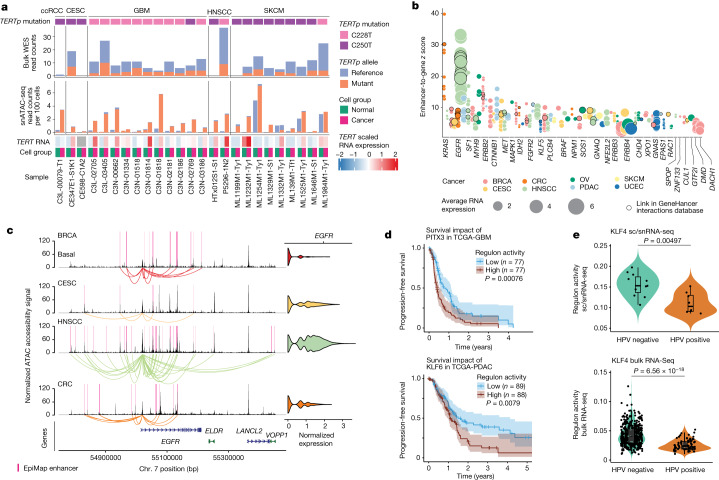
Chromatin accessibility is essential in regulating gene expression and cellular identity, and alterations in accessibility have been implicated in driving cancer initiation, progression and metastasis1-4. Although the genetic contributions to oncogenic transitions have been investigated, epigenetic drivers remain less understood. Here we constructed a pan-cancer epigenetic and transcriptomic atlas using single-nucleus chromatin accessibility data (using single-nucleus assay for transposase-accessible chromatin) from 225 samples and matched single-cell or single-nucleus RNA-sequencing expression data from 206 samples. With over 1 million cells from each platform analysed through the enrichment of accessible chromatin regions, transcription factor motifs and regulons, we identified epigenetic drivers associated with cancer transitions. Some epigenetic drivers appeared in multiple cancers (for example, regulatory regions of ABCC1 and VEGFA; GATA6 and FOX-family motifs), whereas others were cancer specific (for example, regulatory regions of FGF19, ASAP2 and EN1, and the PBX3 motif). Among epigenetically altered pathways, TP53, hypoxia and TNF signalling were linked to cancer initiation, whereas oestrogen response, epithelial-mesenchymal transition and apical junction were tied to metastatic transition. Furthermore, we revealed a marked correlation between enhancer accessibility and gene expression and uncovered cooperation between epigenetic and genetic drivers. This atlas provides a foundation for further investigation of epigenetic dynamics in cancer transitions.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures











References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
