

Emicizumab: the hemophilia A game-changer
- PMID: 37916312
- PMCID: PMC11063855
- DOI: 10.3324/haematol.2022.282099
Emicizumab: the hemophilia A game-changer
Abstract
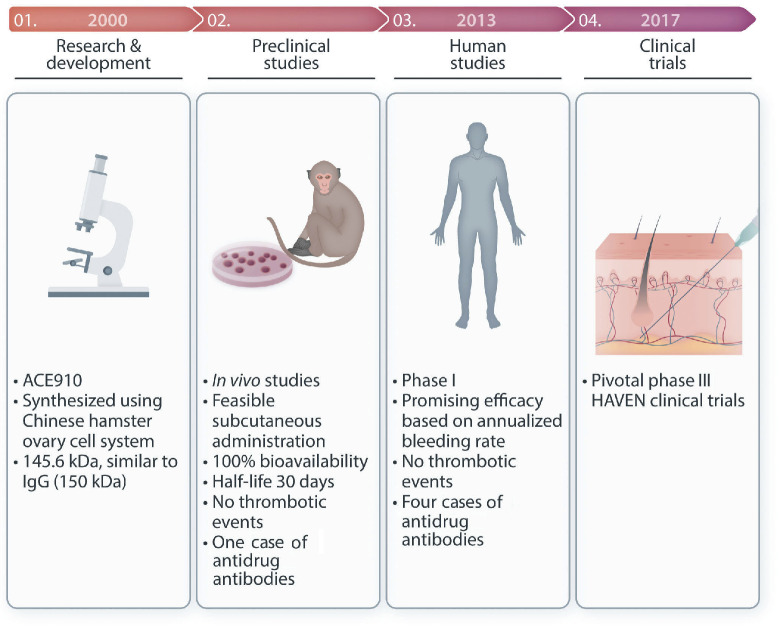
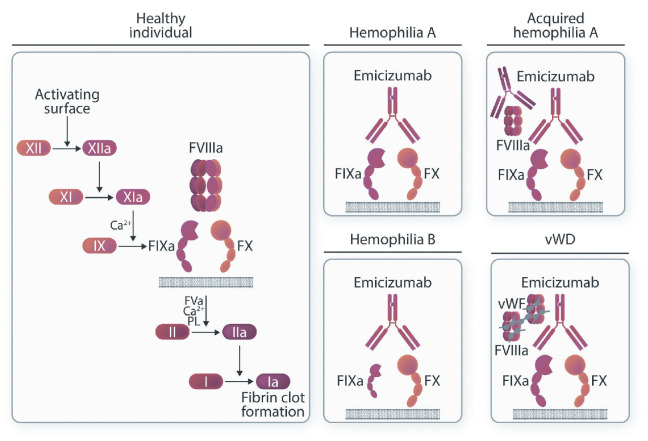
In hemophilia, the unmet needs regarding adherence to prophylaxis and lack of effective long-term prophylaxis regimens, especially in patients with inhibitors, led to the production of emicizumab, the first non-factor medicine for subcutaneous administration in patients with severe and moderate hemophilia A with or without factor VIII inhibitors. This review describes the research steps behind the development of this game-changing medication as well as its success in the prophylaxis of bleeding episodes, as witnessed by the results of pivotal clinical trials but also by real-life use in the frame of a still expanding global market. We also discuss potential and actual adverse events and the nuances related to clinical use, such as laboratory monitoring, development of neutralizing antidrug antibodies, risk of thrombosis/hypercoagulability and role in the management of surgical operations. The potential of emicizumab to prevent bleeding in other congenital and acquired coagulation disorders is also outlined.
Figures
References
-
- Srivastava A, Santagostino E, Dougall A, et al. . WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26 (Suppl 6):1-158. - PubMed
-
- Leissinger C, Gringeri A, Antmen B, et al. . Anti-inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N Engl J Med. 2011;365(18):1684-1692. - PubMed
-
- Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. . Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535-544. - PubMed
-
- Kitazawa T, Shima M. Emicizumab, a humanized bispecific antibody to coagulation factors IXa and X with a factor VIIIacofactor activity. Int J Hematol. 2020;111(1):20-30. - PubMed
-
- Muto A, Yoshihashi K, Takeda M, et al. . Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12(2):206-213. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
