

Dystrophin- and Utrophin-Based Therapeutic Approaches for Treatment of Duchenne Muscular Dystrophy: A Comparative Review
- PMID: 37917377
- PMCID: PMC10789850
- DOI: 10.1007/s40259-023-00632-3
Dystrophin- and Utrophin-Based Therapeutic Approaches for Treatment of Duchenne Muscular Dystrophy: A Comparative Review
Erratum in
-
Correction: Dystrophin- and Utrophin-Based Therapeutic Approaches for Treatment of Duchenne Muscular Dystrophy: A Comparative Review.BioDrugs. 2025 Jan;39(1):167. doi: 10.1007/s40259-024-00693-y. BioDrugs. 2025. PMID: 39786649 Free PMC article. No abstract available.
Abstract
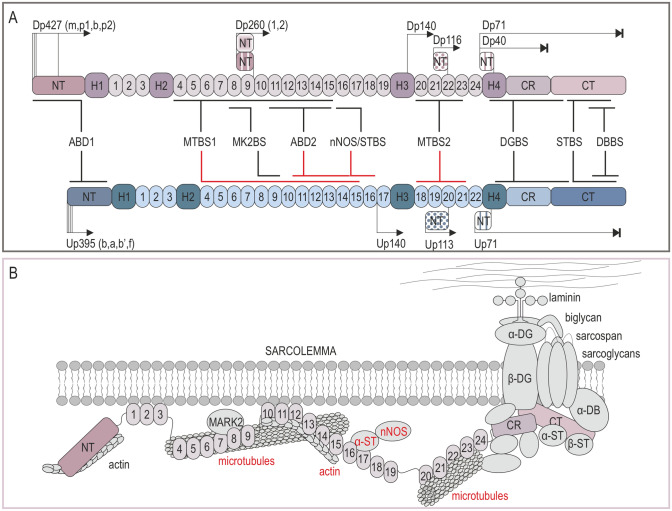
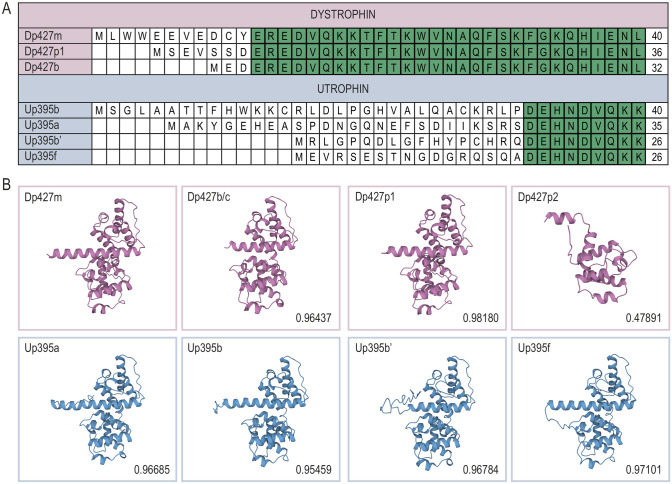
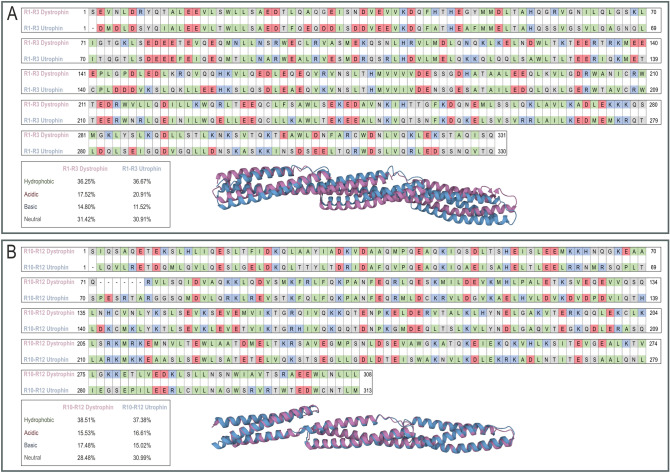
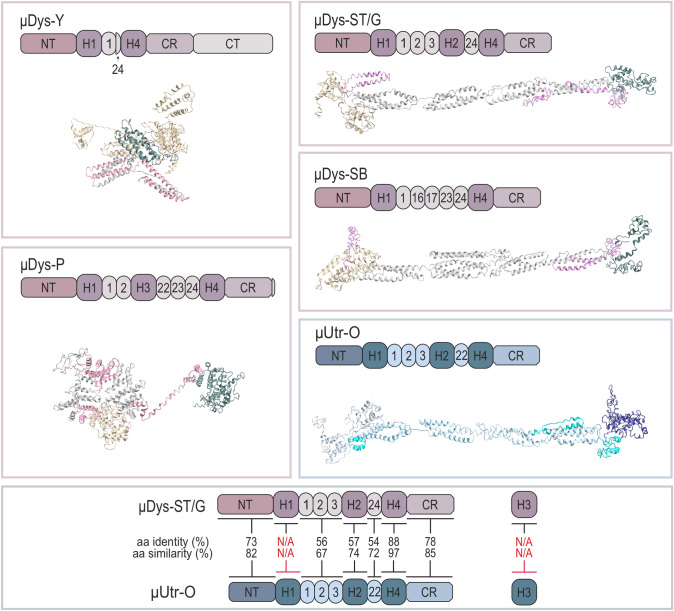
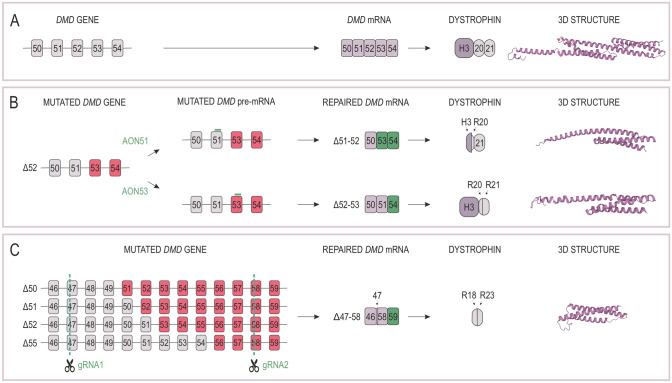
Duchenne muscular dystrophy is a devastating disease that leads to progressive muscle loss and premature death. While medical management focuses mostly on symptomatic treatment, decades of research have resulted in first therapeutics able to restore the affected reading frame of dystrophin transcripts or induce synthesis of a truncated dystrophin protein from a vector, with other strategies based on gene therapy and cell signaling in preclinical or clinical development. Nevertheless, recent reports show that potentially therapeutic dystrophins can be immunogenic in patients. This raises the question of whether a dystrophin paralog, utrophin, could be a more suitable therapeutic protein. Here, we compare dystrophin and utrophin amino acid sequences and structures, combining published data with our extended in silico analyses. We then discuss these results in the context of therapeutic approaches for Duchenne muscular dystrophy. Specifically, we focus on strategies based on delivery of micro-dystrophin and micro-utrophin genes with recombinant adeno-associated viral vectors, exon skipping of the mutated dystrophin pre-mRNAs, reading through termination codons with small molecules that mask premature stop codons, dystrophin gene repair by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (CRISPR/Cas9)-mediated genetic engineering, and increasing utrophin levels. Our analyses highlight the importance of various dystrophin and utrophin domains in Duchenne muscular dystrophy treatment, providing insights into designing novel therapeutic compounds with improved efficacy and decreased immunoreactivity. While the necessary actin and β-dystroglycan binding sites are present in both proteins, important functional distinctions can be identified in these domains and some other parts of truncated dystrophins might need redesigning due to their potentially immunogenic qualities. Alternatively, therapies based on utrophins might provide a safer and more effective approach.
© 2023. The Author(s).
Conflict of interest statement
JSC is one of the inventors on patents covering muscle-specific gene editing and patents covering muscle-specific gene regulatory cassettes, micro-dystrophins, and systemic delivery of AAV. JSC holds equity in and is a member of the scientific advisory board of Solid Biosciences. PK, SS, and ZK have no competing interests to declare.
Figures


References
-
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003. 10.1016/s1474-4422(03)00585-4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
