

Deciphering the molecular and cellular atlas of immune cells in septic patients with different bacterial infections
- PMID: 37919720
- PMCID: PMC10621118
- DOI: 10.1186/s12967-023-04631-4
Deciphering the molecular and cellular atlas of immune cells in septic patients with different bacterial infections
Abstract
Background: Sepsis is a life-threatening organ dysfunction caused by abnormal immune responses to various, predominantly bacterial, infections. Different bacterial infections lead to substantial variation in disease manifestation and therapeutic strategies. However, the underlying cellular heterogeneity and mechanisms involved remain poorly understood.
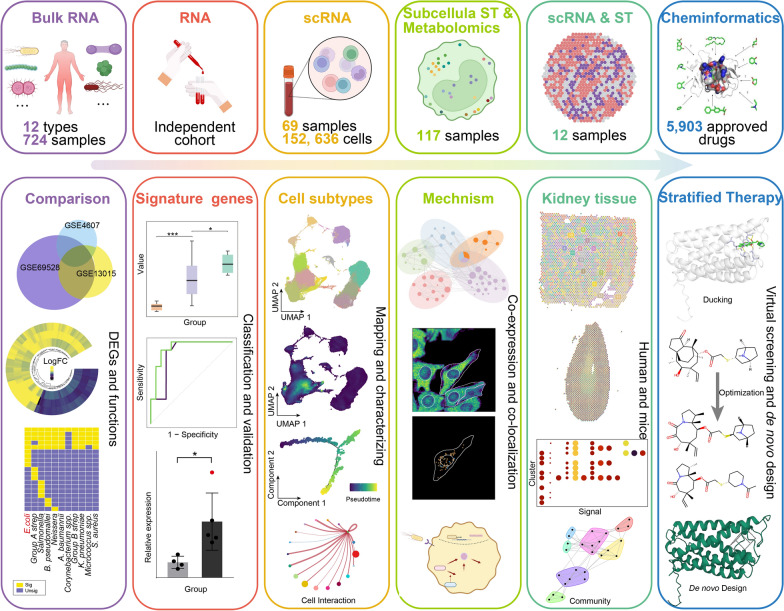
Methods: Multiple bulk transcriptome datasets from septic patients with 12 types of bacterial infections were integrated to identify signature genes for each infection. Signature genes were mapped onto an integrated large single-cell RNA (scRNA) dataset from septic patients, to identify subsets of cells associated with different sepsis types, and multiple omics datasets were combined to reveal the underlying molecular mechanisms. In addition, an scRNA dataset and spatial transcriptome data were used to identify signaling pathways in sepsis-related cells. Finally, molecular screening, optimization, and de novo design were conducted to identify potential targeted drugs and compounds.
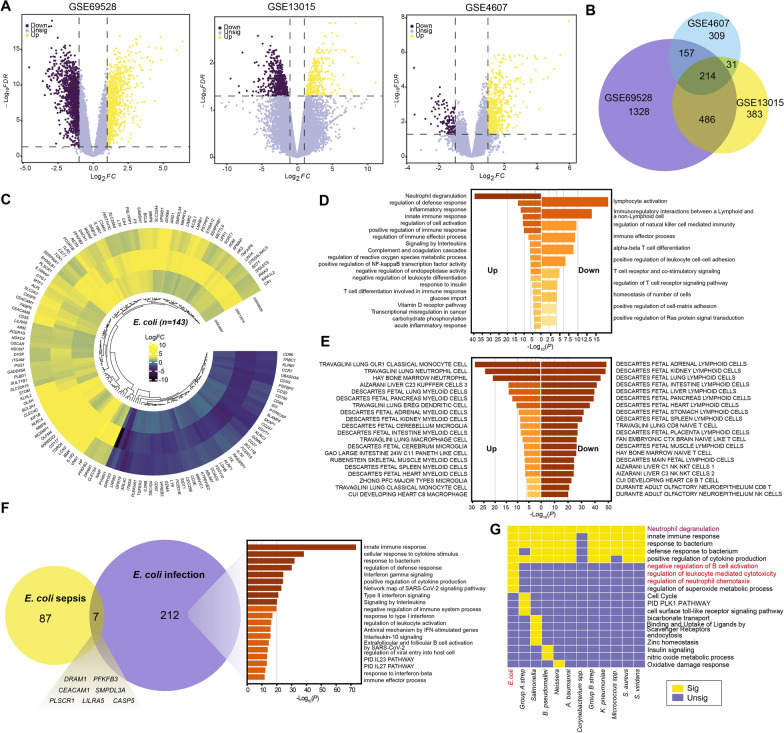
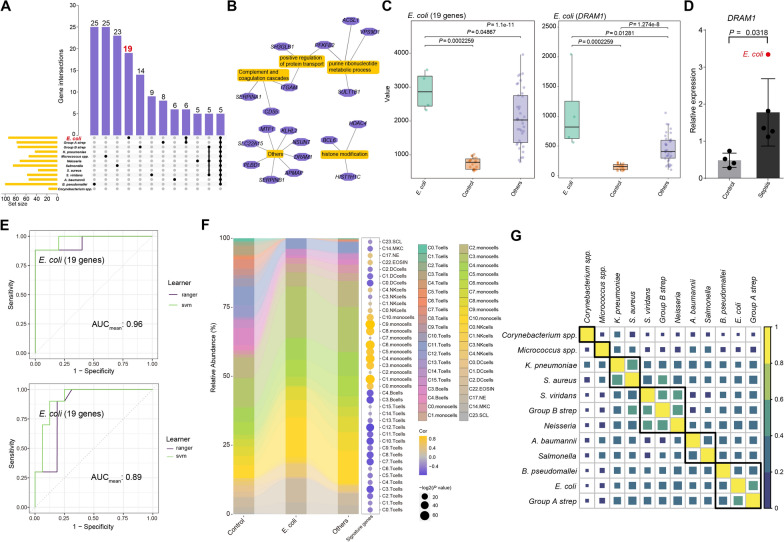
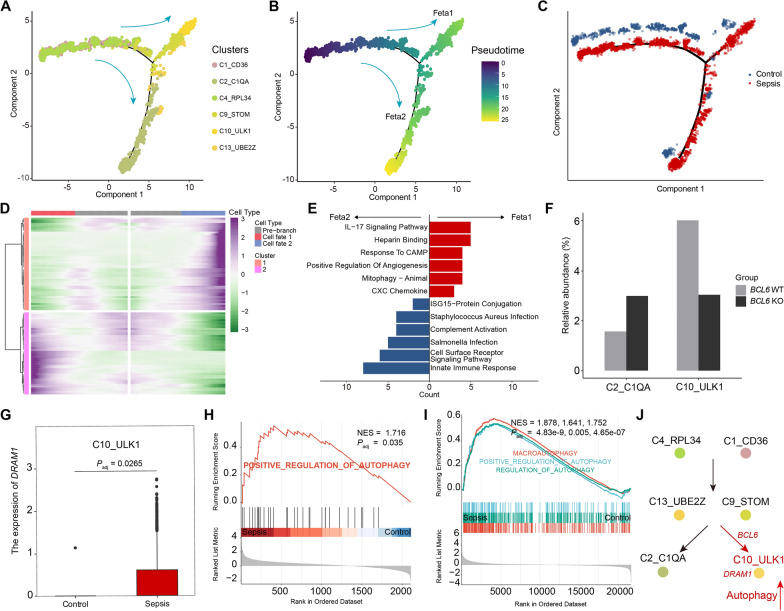
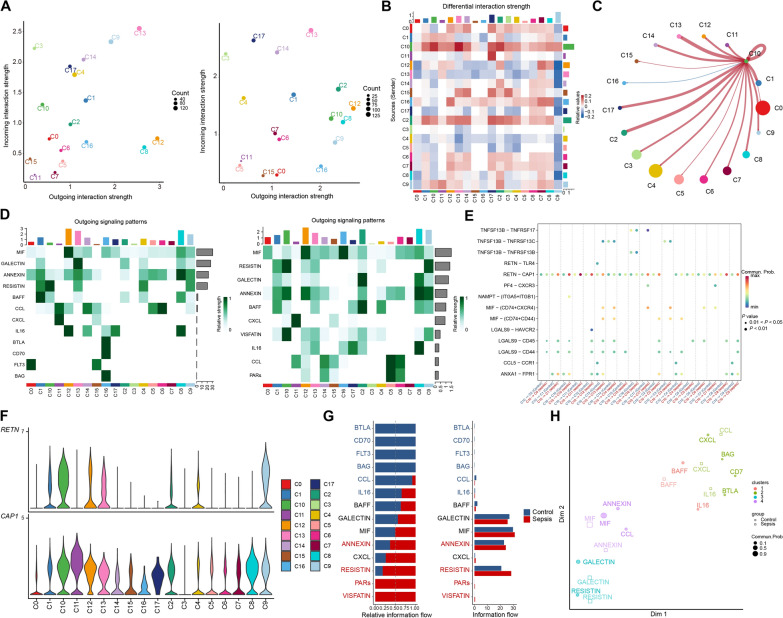
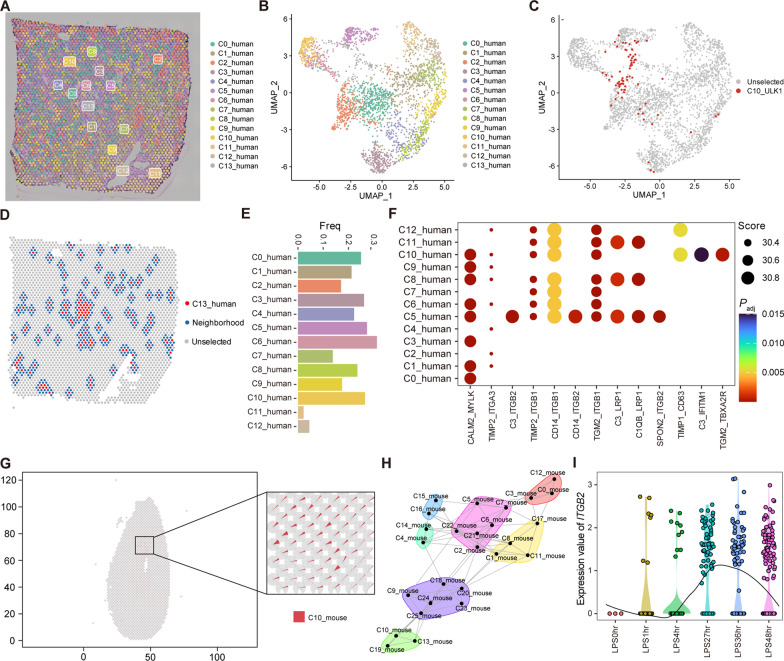
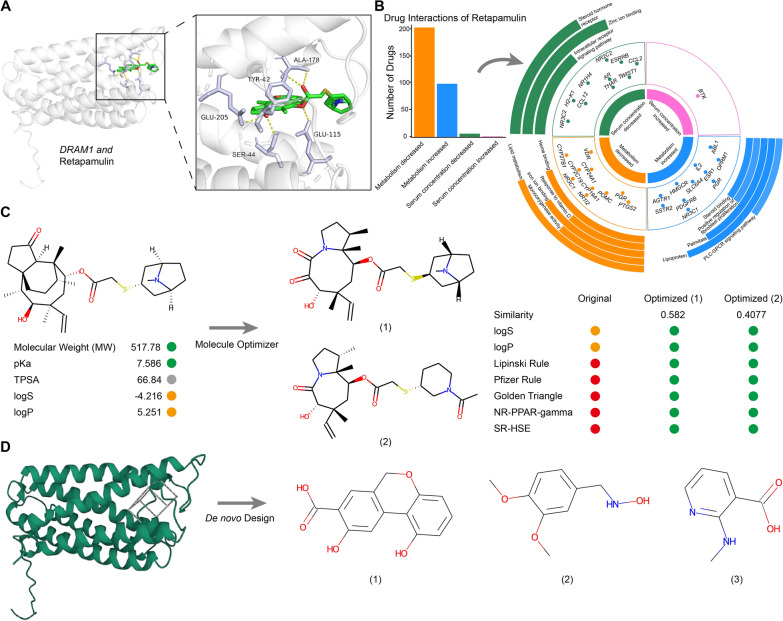
Results: We elucidated the cellular heterogeneity among septic patients with different bacterial infections. In Escherichia coli (E. coli) sepsis, 19 signature genes involved in epigenetic regulation and metabolism were identified, of which DRAM1 was demonstrated to promote autophagy and glycolysis in response to E. coli infection. DRAM1 upregulation was confirmed in an independent sepsis cohort. Further, we showed that DRAM1 could maintain survival of a pro-inflammatory monocyte subset, C10_ULK1, which induces systemic inflammation by interacting with other cell subsets via resistin and integrin signaling pathways in blood and kidney tissue, respectively. Finally, retapamulin was identified and optimized as a potential drug for treatment of E. coli sepsis targeting the signature gene, DRAM1, and inhibiting E. coli protein synthesis. Several other targeted drugs were also identified in other types of sepsis, including nystatin targeting C1QA in Neisseria sepsis and dalfopristin targeting CTSD in Streptococcus viridans sepsis.
Conclusion: Our study provides a comprehensive overview of the cellular heterogeneity and underlying mechanisms in septic patients with various bacterial infections, providing insights to inform development of stratified targeted therapies for sepsis.
Keywords: Cellular heterogeneity; Multi-omics; Sepsis; Stratified targeted therapies; Various bacterial infections.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
