

Multi-scale characterisation of homologous recombination deficiency in breast cancer
- PMID: 37919776
- PMCID: PMC10621207
- DOI: 10.1186/s13073-023-01239-7
Multi-scale characterisation of homologous recombination deficiency in breast cancer
Abstract
Background: Homologous recombination is a robust, broadly error-free mechanism of double-strand break repair, and deficiencies lead to PARP inhibitor sensitivity. Patients displaying homologous recombination deficiency can be identified using 'mutational signatures'. However, these patterns are difficult to reliably infer from exome sequencing. Additionally, as mutational signatures are a historical record of mutagenic processes, this limits their utility in describing the current status of a tumour.
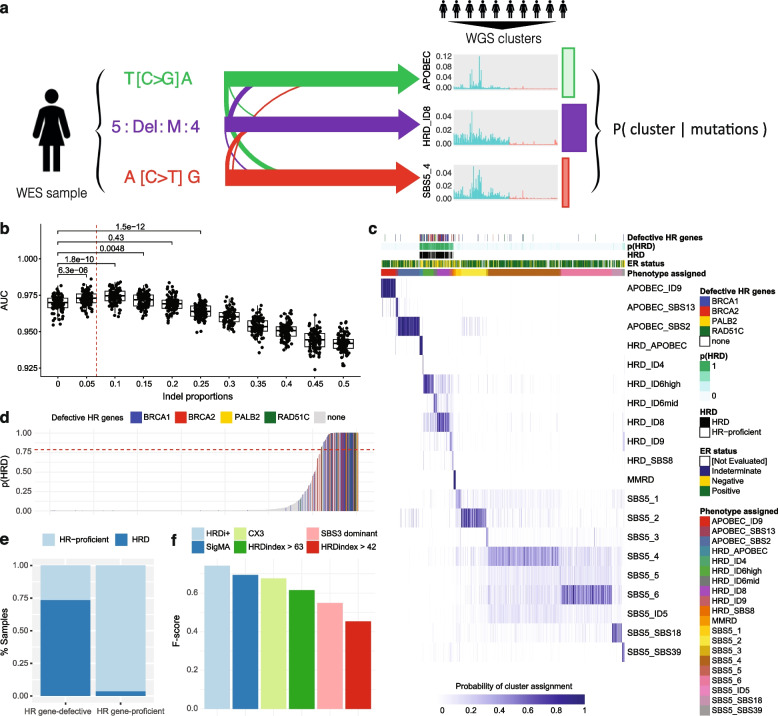
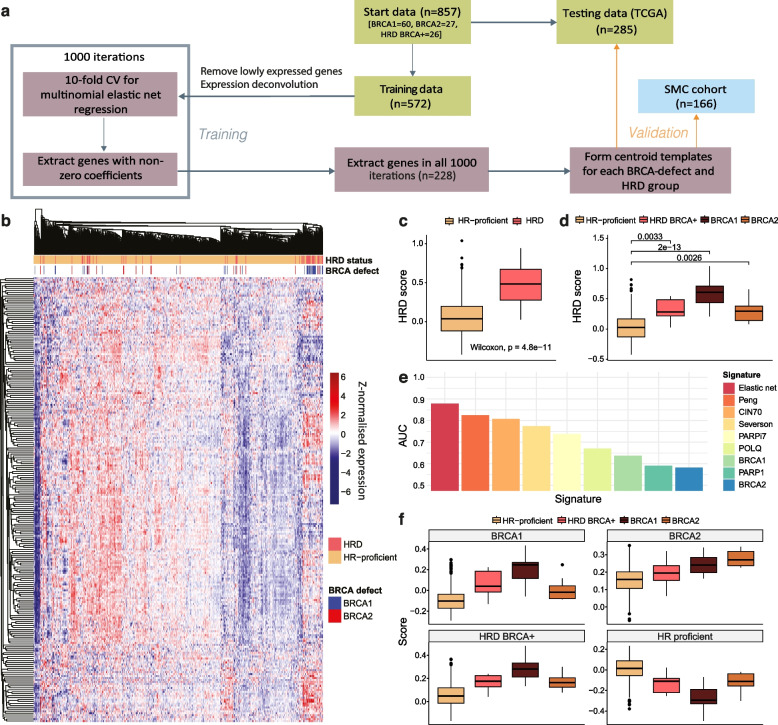
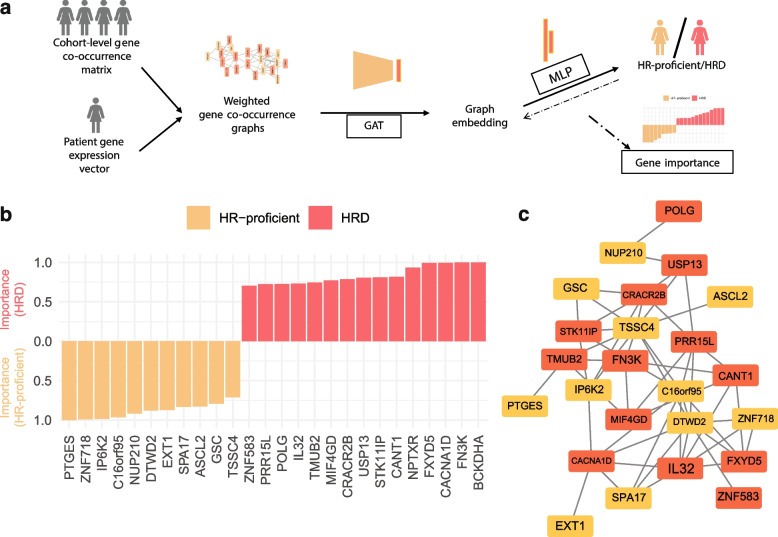
Methods: We apply two methods for characterising homologous recombination deficiency in breast cancer to explore the features and heterogeneity associated with this phenotype. We develop a likelihood-based method which leverages small insertions and deletions for high-confidence classification of homologous recombination deficiency for exome-sequenced breast cancers. We then use multinomial elastic net regression modelling to develop a transcriptional signature of heterogeneous homologous recombination deficiency. This signature is then applied to single-cell RNA-sequenced breast cancer cohorts enabling analysis of homologous recombination deficiency heterogeneity and differential patterns of tumour microenvironment interactivity.
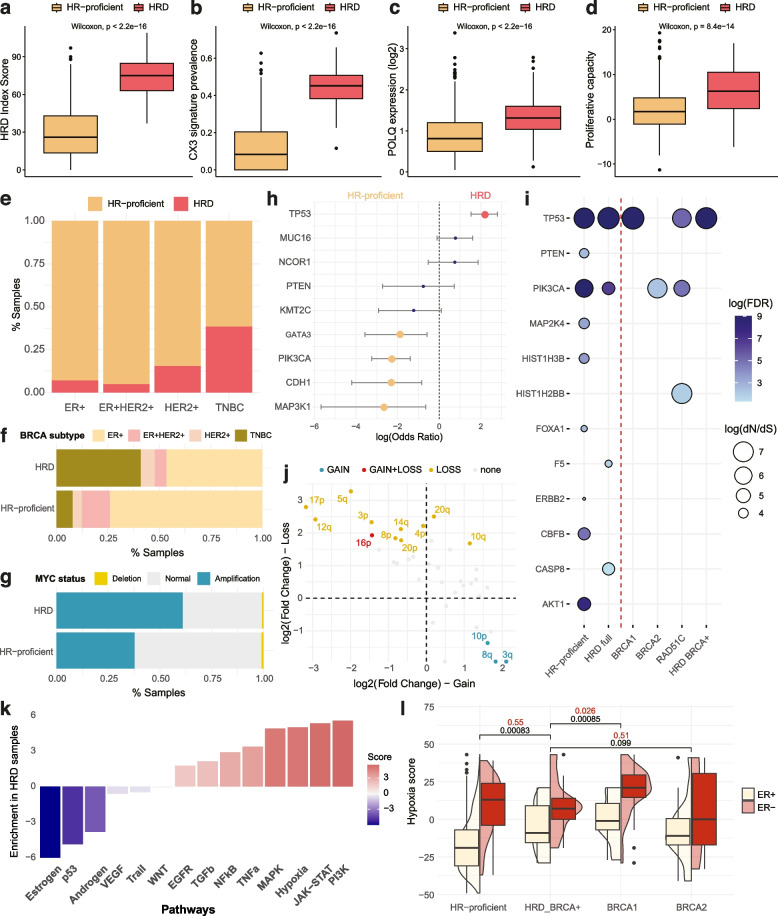
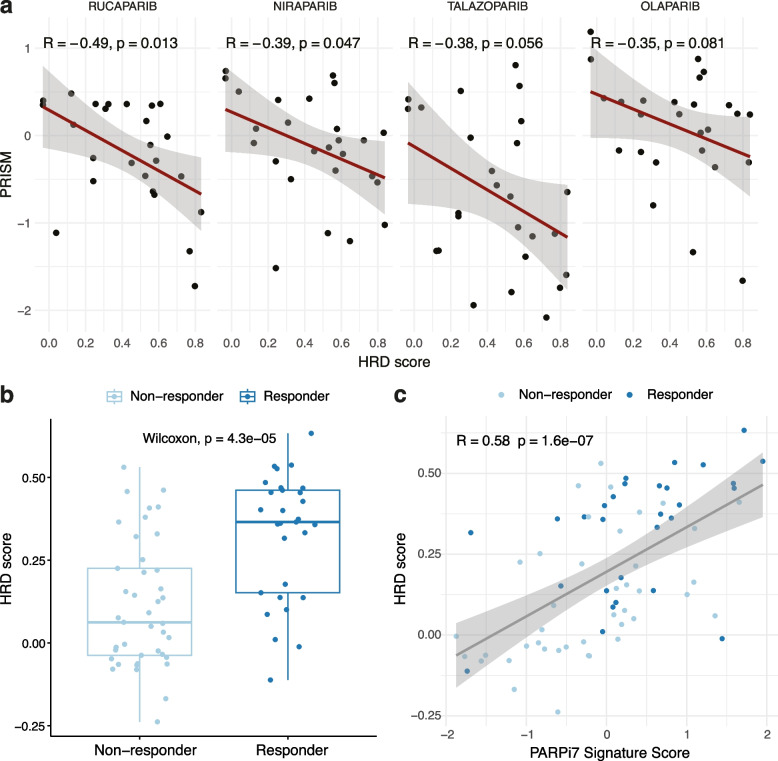
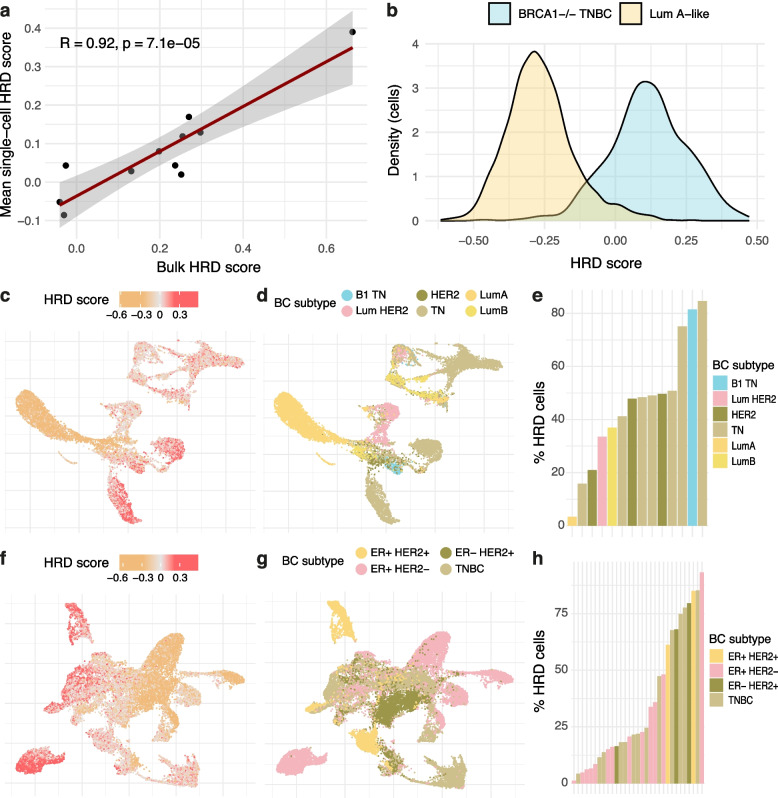
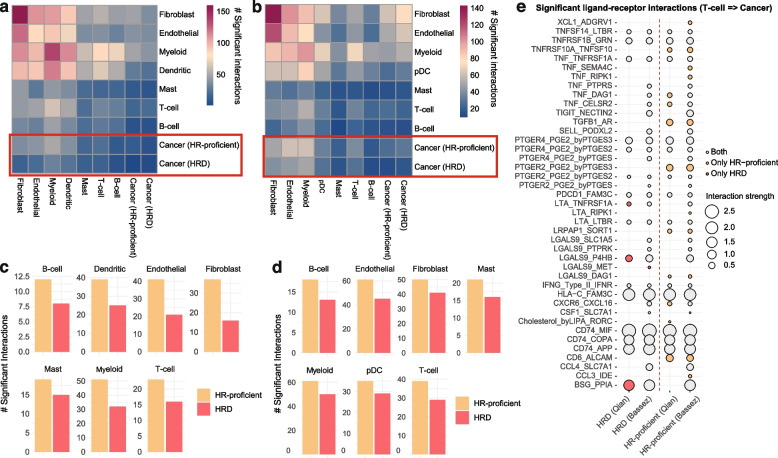
Results: We demonstrate that the inclusion of indel events, even at low levels, improves homologous recombination deficiency classification. Whilst BRCA-positive homologous recombination deficient samples display strong similarities to those harbouring BRCA1/2 defects, they appear to deviate in microenvironmental features such as hypoxic signalling. We then present a 228-gene transcriptional signature which simultaneously characterises homologous recombination deficiency and BRCA1/2-defect status, and is associated with PARP inhibitor response. Finally, we show that this signature is applicable to single-cell transcriptomics data and predict that these cells present a distinct milieu of interactions with their microenvironment compared to their homologous recombination proficient counterparts, typified by a decreased cancer cell response to TNFα signalling.
Conclusions: We apply multi-scale approaches to characterise homologous recombination deficiency in breast cancer through the development of mutational and transcriptional signatures. We demonstrate how indels can improve homologous recombination deficiency classification in exome-sequenced breast cancers. Additionally, we demonstrate the heterogeneity of homologous recombination deficiency, especially in relation to BRCA1/2-defect status, and show that indications of this feature can be captured at a single-cell level, enabling further investigations into interactions between DNA repair deficient cells and their tumour microenvironment.
Keywords: Breast cancer; Homologous recombination deficiency; Mutational signatures; PARP inhibition; Single cell; Transcriptional classifier; Tumour microenvironment.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
