

Synthesis, crystal structure, DFT, Hirshfeld surface analysis, energy frameworks and in-Silico drug-targeting PFKFB3 kinase of novel triazolequinoxalin derivative (TZQ) as a therapeutic Strategy against cancer
- PMID: 37920528
- PMCID: PMC10618769
- DOI: 10.1016/j.heliyon.2023.e21312
Synthesis, crystal structure, DFT, Hirshfeld surface analysis, energy frameworks and in-Silico drug-targeting PFKFB3 kinase of novel triazolequinoxalin derivative (TZQ) as a therapeutic Strategy against cancer
Abstract
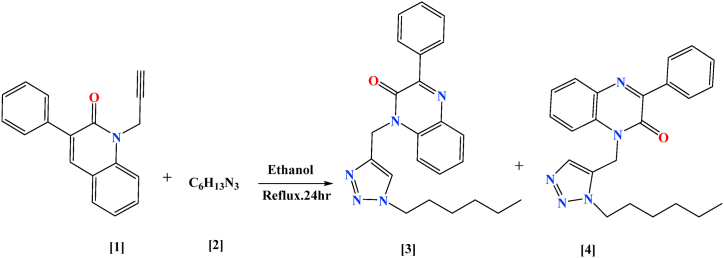
Overall, drug design is a dynamic and evolving field, with researchers constantly working to improve their understanding of molecular interactions, develop new computational methods, and explore innovative techniques for creating effective and safe medications. The process can involve steps such as the identification of targets, the discovery of lead compounds, lead optimization, preliminary testing, human trials, regulatory approval and finally post-marketing surveillance, all aimed at bringing a new drug from concept to market. In this article, the synthesis of the novel triazolequinoxalin (TZQ) 1-((1-hexyl-1H-1,2,3-triazol-5-yl)methyl)-3-phenylquinoxalin-2(1H)-one (4) is reported. The structure has been identified with a variety of spectroscopic methods (1H, 13C NMR, and LC-MS) and finally, the structure has been determined by X-ray diffraction (XRD) studies. The TZQ molecule has crystallized in the monoclinic space C2/c group with unit cell dimensions a = 41.201(2) Å, b = 10.6339(6) Å, c = 9.4997(4) Å, β = 93.904(4). The crystal structure is stabilized by intermolecular interactions (N-H ⋯ O and N-H … Cg) occurring within the molecule. The presence of these intermolecular interactions is evaluated through analysis of Hirshfeld surfaces (HS) and two-dimensional (2D) chemical fingerprints map. Additionally, energy frameworks were employed to identify the prevailing interaction energy influencing the molecular arrangement. Density Functional Theory (DFT) calculations were computed to establish concurrence between theoretical and experimental results. Furthermore, the HOMO-LUMO energy levels were determined using the B3LYP/6-31+G(d, p) level of theory. Finally, molecular docking was used to predict the anti-cancer activity of the compound (4) against PFKFB3 kinase and presented noticeable hydrophilic and hydrophobic interactions at the active site region.
Keywords: Anti-cancer docking studies; Energy framework; Hirshfeld surface; Triazolequinoxalin.
© 2023 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures



 O⋅⋅⋅π interactions. Symmetry codes: (i) x, y, −1+z; (ii) 3/2- x, −1/2+ y, 1/2- z, (iii) x, -y, ½+z.
O⋅⋅⋅π interactions. Symmetry codes: (i) x, y, −1+z; (ii) 3/2- x, −1/2+ y, 1/2- z, (iii) x, -y, ½+z.





References
-
- Zabiulla, et al. Recent investigation on heterocycles with one nitrogen [piperidine, pyridine and quinoline], two nitrogen [1, 3, 4-thiadiazole and pyrazole] and three nitrogen [1, 2, 4-triazole]: a review. J. Iran. Chem. Soc. 2022;19(1):23–54. doi: 10.1007/s13738-021-02293-x. - DOI
-
- Al-Ostoot F.H., et al. Recent investigations into synthesis and pharmacological activities of phenoxy acetamide and its derivatives (chalcone, indole and quinoline) as possible therapeutic candidates. J. Iran. Chem. Soc. 2021;18:1839–1875. doi: 10.1007/s13738-021-02172-5. - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous
