

Blockage of EGFR/AKT and mevalonate pathways synergize the antitumor effect of temozolomide by reprogramming energy metabolism in glioblastoma
- PMID: 37920878
- PMCID: PMC10693308
- DOI: 10.1002/cac2.12502
Blockage of EGFR/AKT and mevalonate pathways synergize the antitumor effect of temozolomide by reprogramming energy metabolism in glioblastoma
Abstract
Background: Metabolism reprogramming plays a vital role in glioblastoma (GBM) progression and recurrence by producing enough energy for highly proliferating tumor cells. In addition, metabolic reprogramming is crucial for tumor growth and immune-escape mechanisms. Epidermal growth factor receptor (EGFR) amplification and EGFR-vIII mutation are often detected in GBM cells, contributing to the malignant behavior. This study aimed to investigate the functional role of the EGFR pathway on fatty acid metabolism remodeling and energy generation.
Methods: Clinical GBM specimens were selected for single-cell RNA sequencing and untargeted metabolomics analysis. A metabolism-associated RTK-fatty acid-gene signature was constructed and verified. MK-2206 and MK-803 were utilized to block the RTK pathway and mevalonate pathway induced abnormal metabolism. Energy metabolism in GBM with activated EGFR pathway was monitored. The antitumor effect of Osimertinib and Atorvastatin assisted by temozolomide (TMZ) was analyzed by an intracranial tumor model in vivo.
Results: GBM with high EGFR expression had characteristics of lipid remodeling and maintaining high cholesterol levels, supported by the single-cell RNA sequencing and metabolomics of clinical GBM samples. Inhibition of the EGFR/AKT and mevalonate pathways could remodel energy metabolism by repressing the tricarboxylic acid cycle and modulating ATP production. Mechanistically, the EGFR/AKT pathway upregulated the expressions of acyl-CoA synthetase short-chain family member 3 (ACSS3), acyl-CoA synthetase long-chain family member 3 (ACSL3), and long-chain fatty acid elongation-related gene ELOVL fatty acid elongase 2 (ELOVL2) in an NF-κB-dependent manner. Moreover, inhibition of the mevalonate pathway reduced the EGFR level on the cell membranes, thereby affecting the signal transduction of the EGFR/AKT pathway. Therefore, targeting the EGFR/AKT and mevalonate pathways enhanced the antitumor effect of TMZ in GBM cells and animal models.
Conclusions: Our findings not only uncovered the mechanism of metabolic reprogramming in EGFR-activated GBM but also provided a combinatorial therapeutic strategy for clinical GBM management.
Keywords: EGFR; combinatorial therapeutic strategy; energy metabolism; glioblastoma.
© 2023 The Authors. Cancer Communications published by John Wiley & Sons Australia, Ltd on behalf of SUN YAT-SEN UNIVERSITY CANCER CENTER.
Conflict of interest statement
The authors declare that there is no conflict of interest in this study.
Figures






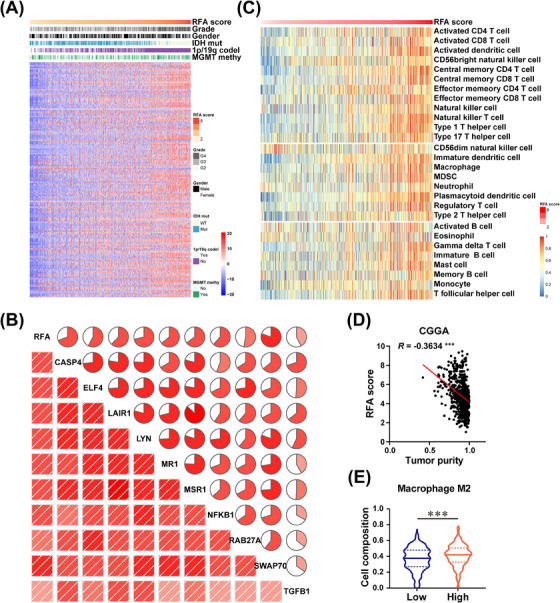



References
-
- Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829–848. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 82002657/National Natural Science Foundation of China
- 82073322/National Natural Science Foundation of China
- 81761168038/National Natural Science Foundation of China
- H2020201206/Hebei Natural Science Foundation Precision Medicine Joint Project
- 20YFZCSY00360/Tianjin Key R&D Plan of Tianjin Science and Technology Plan Project
- Brain Tumor Precision Diagnosis and Treatment and Translational Medicine Innovation Unit
- 2019-I2M-5-021/Chinese Academy of Medical Sciences
- TJWJ2021QN003/Science and Technology Project of Tianjin Municipal Health Commission
- 2023B1111020008/Key-Area Research and Development Program of Guangdong Province
- 21JCQNJC01250/Natural Science Foundation of Tianjin Municipal Science and Technology Commission
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
