

Functional screening of amplification outlier oncogenes in organoid models of early tumorigenesis
- PMID: 37922313
- PMCID: PMC10841581
- DOI: 10.1016/j.celrep.2023.113355
Functional screening of amplification outlier oncogenes in organoid models of early tumorigenesis
Abstract
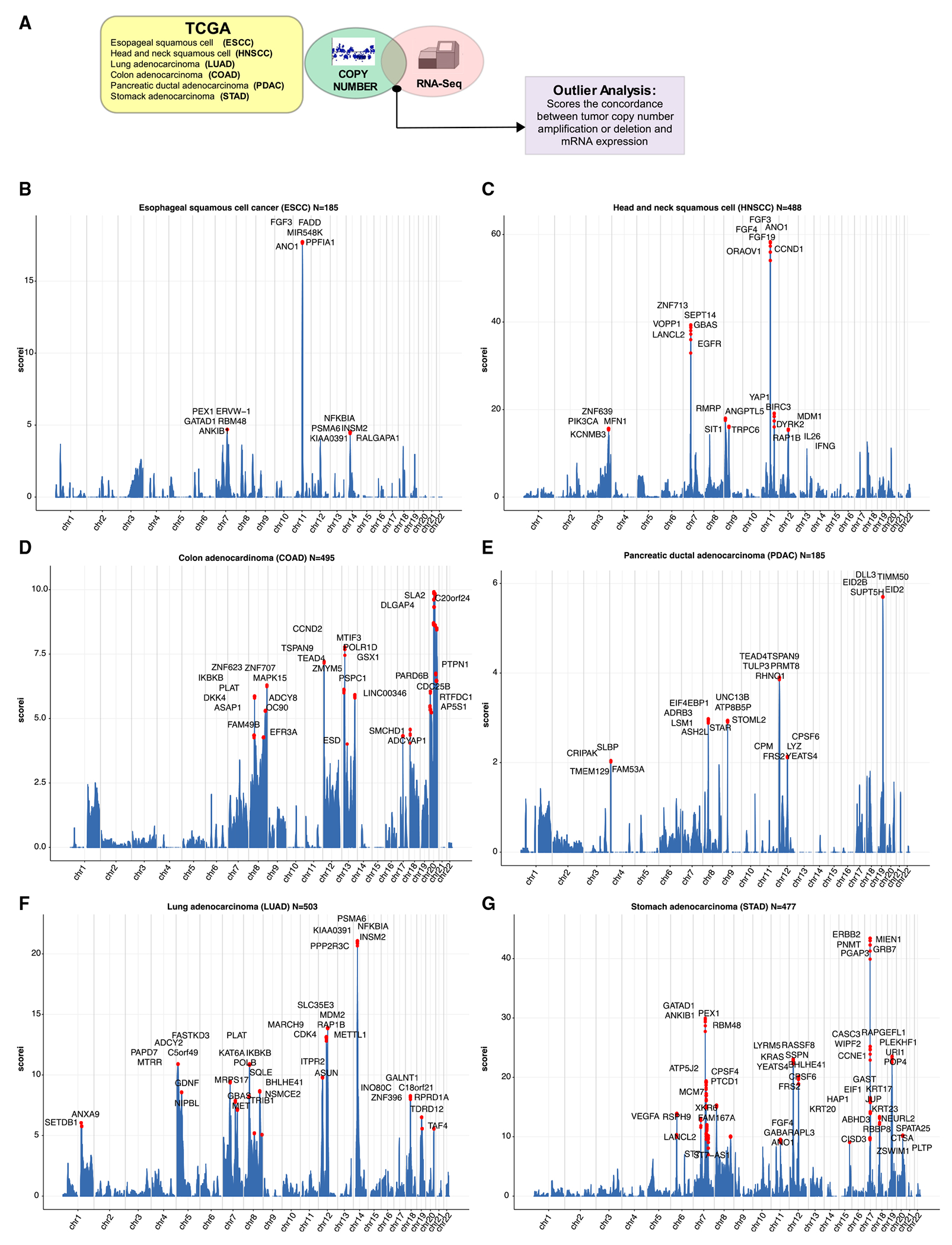
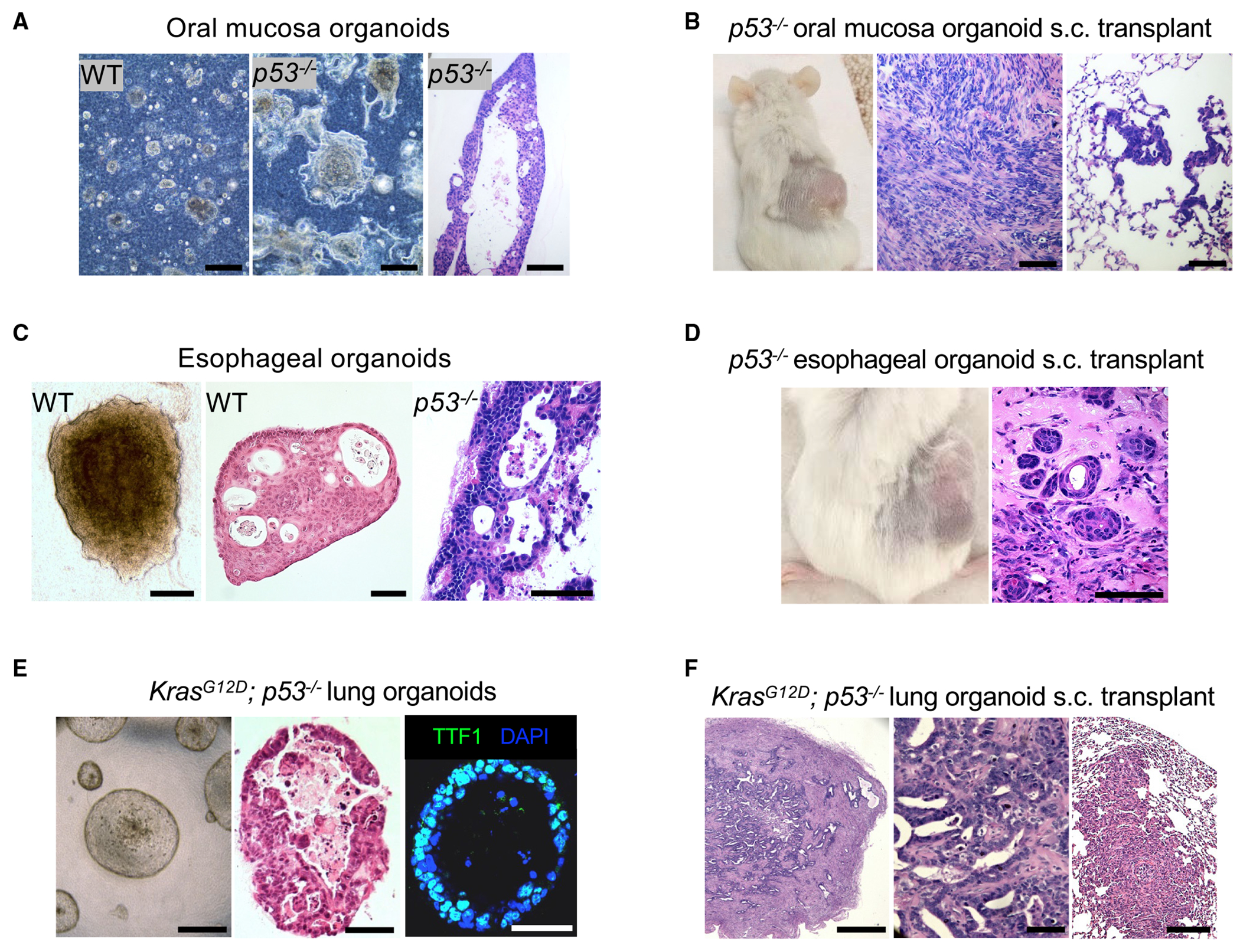
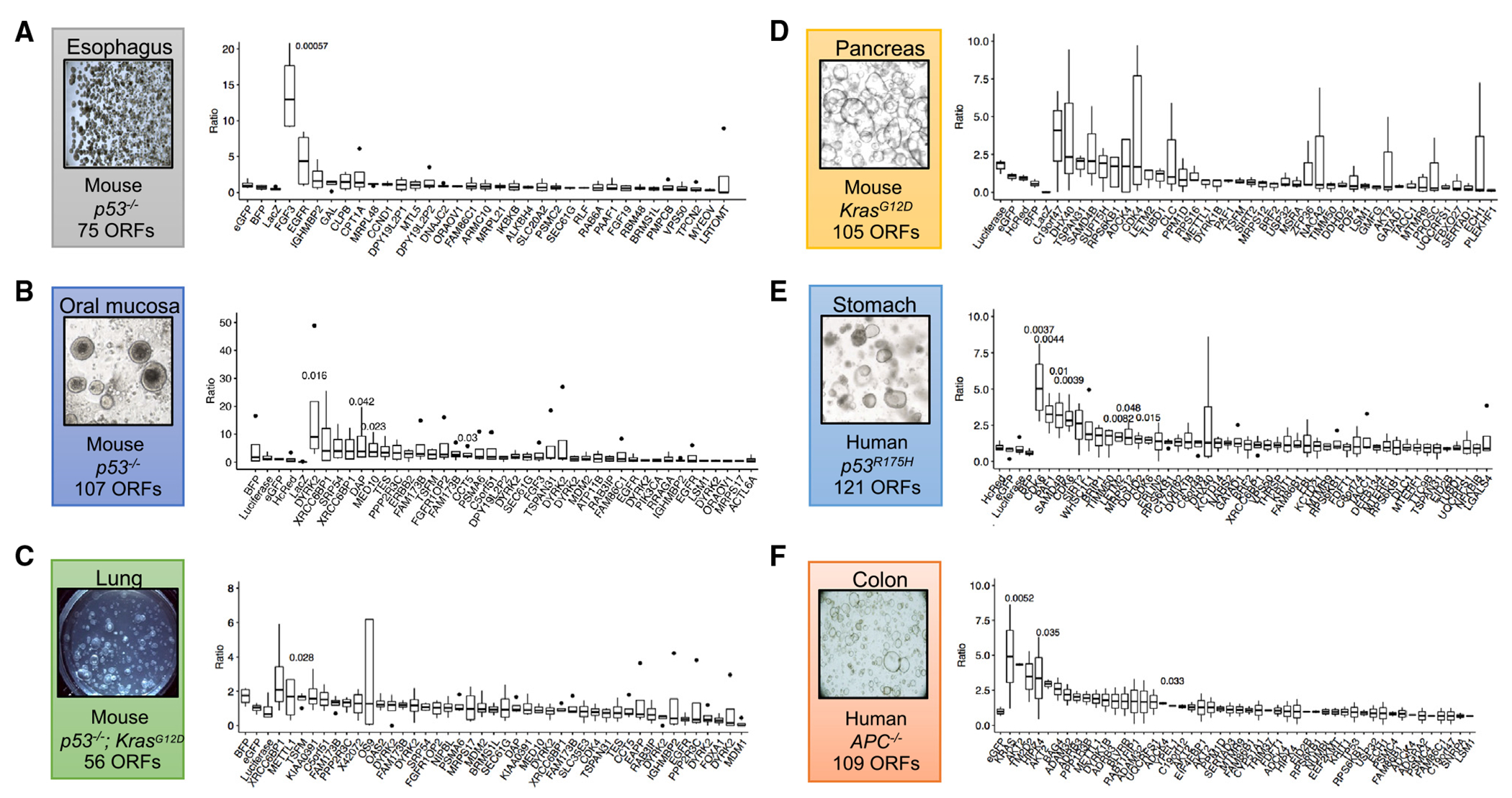
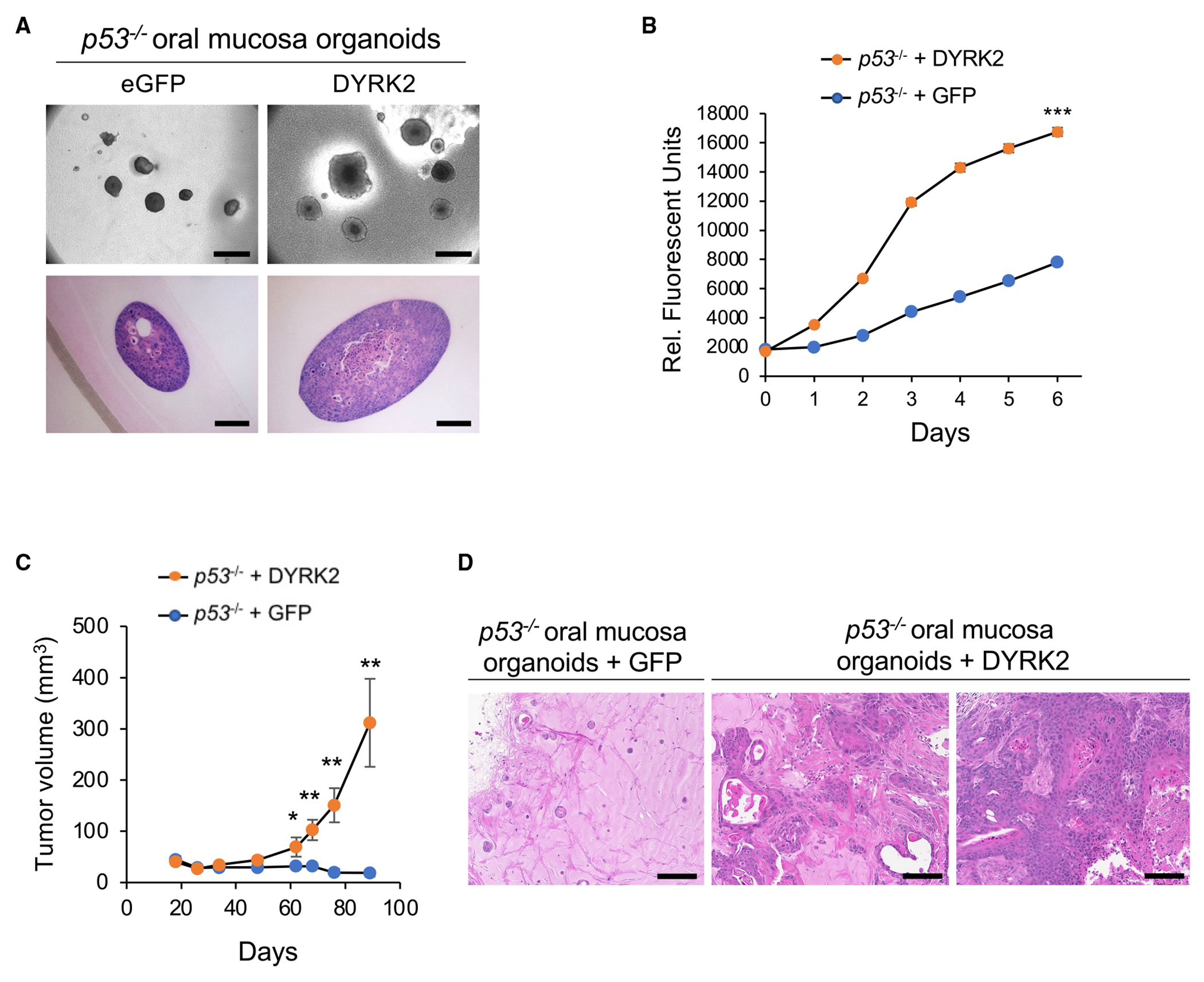
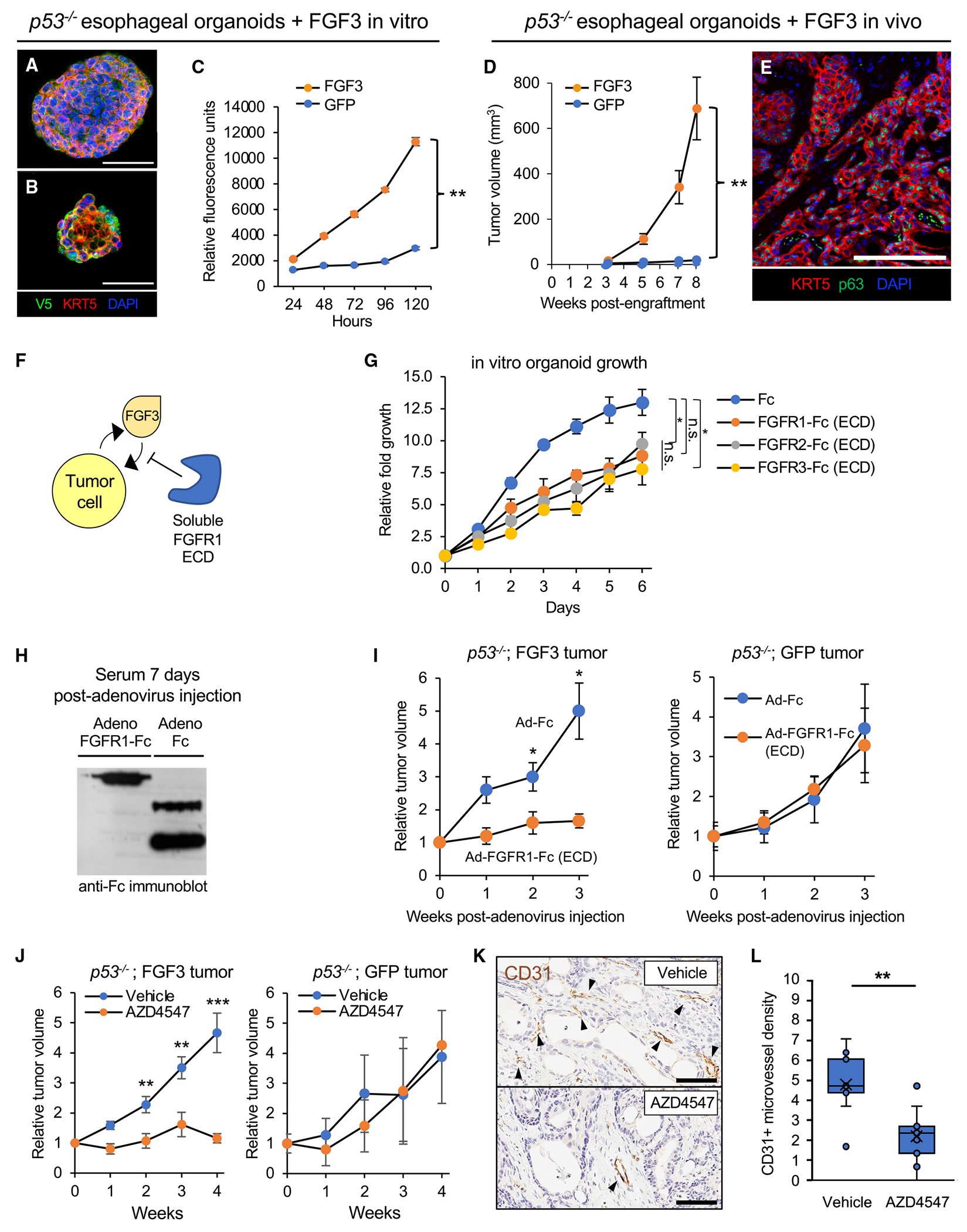
Somatic copy number gains are pervasive across cancer types, yet their roles in oncogenesis are insufficiently evaluated. This inadequacy is partly due to copy gains spanning large chromosomal regions, obscuring causal loci. Here, we employed organoid modeling to evaluate candidate oncogenic loci identified via integrative computational analysis of extreme copy gains overlapping with extreme expression dysregulation in The Cancer Genome Atlas. Subsets of "outlier" candidates were contextually screened as tissue-specific cDNA lentiviral libraries within cognate esophagus, oral cavity, colon, stomach, pancreas, and lung organoids bearing initial oncogenic mutations. Iterative analysis nominated the kinase DYRK2 at 12q15 as an amplified head and neck squamous carcinoma oncogene in p53-/- oral mucosal organoids. Similarly, FGF3, amplified at 11q13 in 41% of esophageal squamous carcinomas, promoted p53-/- esophageal organoid growth reversible by small molecule and soluble receptor antagonism of FGFRs. Our studies establish organoid-based contextual screening of candidate genomic drivers, enabling functional evaluation during early tumorigenesis.
Keywords: CP: Cancer; DYRK2; FGF3; amplification; cancer; cancer driver; copy number alteration; functional genomics; organoid; precision oncology; squamous cancer.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.A.S. has served as a consultant for Boehringer Ingelheim and Pharmacosmos and is a former employee of Tempus Labs. C.J.K. is a scientific advisory board member for Surrozen, Inc., Mozart Therapeutics, and NextVivo. C.C. has served as a scientific advisory board member/consultant for Genentech, Grail, DeepCell, Nanostring, and Viosera. W.C.H. is a consultant for Thermo Fisher, Solasta Ventures, MPM Capital, KSQ Therapeutics, Tyra Biosciences, Frontier Medicine, Jubilant Therapeutics, RAPPTA Therapeutics, Hexagon Bio, Serinus Biosciences, Function Oncology, and Calyx.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
