

Single-cell epigenetic, transcriptional, and protein profiling of latent and active HIV-1 reservoir revealed that IKZF3 promotes HIV-1 persistence
- PMID: 37922905
- PMCID: PMC10843106
- DOI: 10.1016/j.immuni.2023.10.002
Single-cell epigenetic, transcriptional, and protein profiling of latent and active HIV-1 reservoir revealed that IKZF3 promotes HIV-1 persistence
Abstract
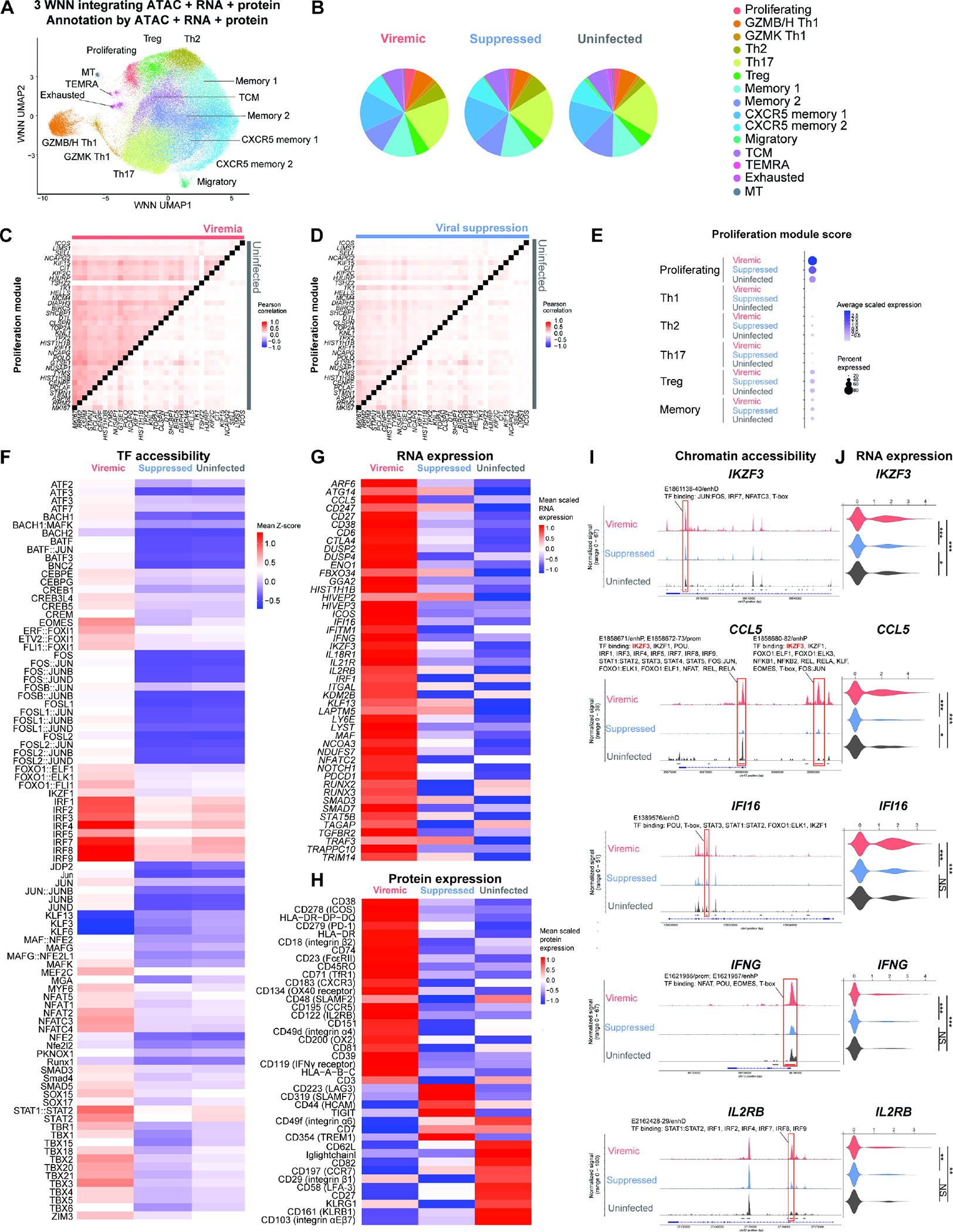
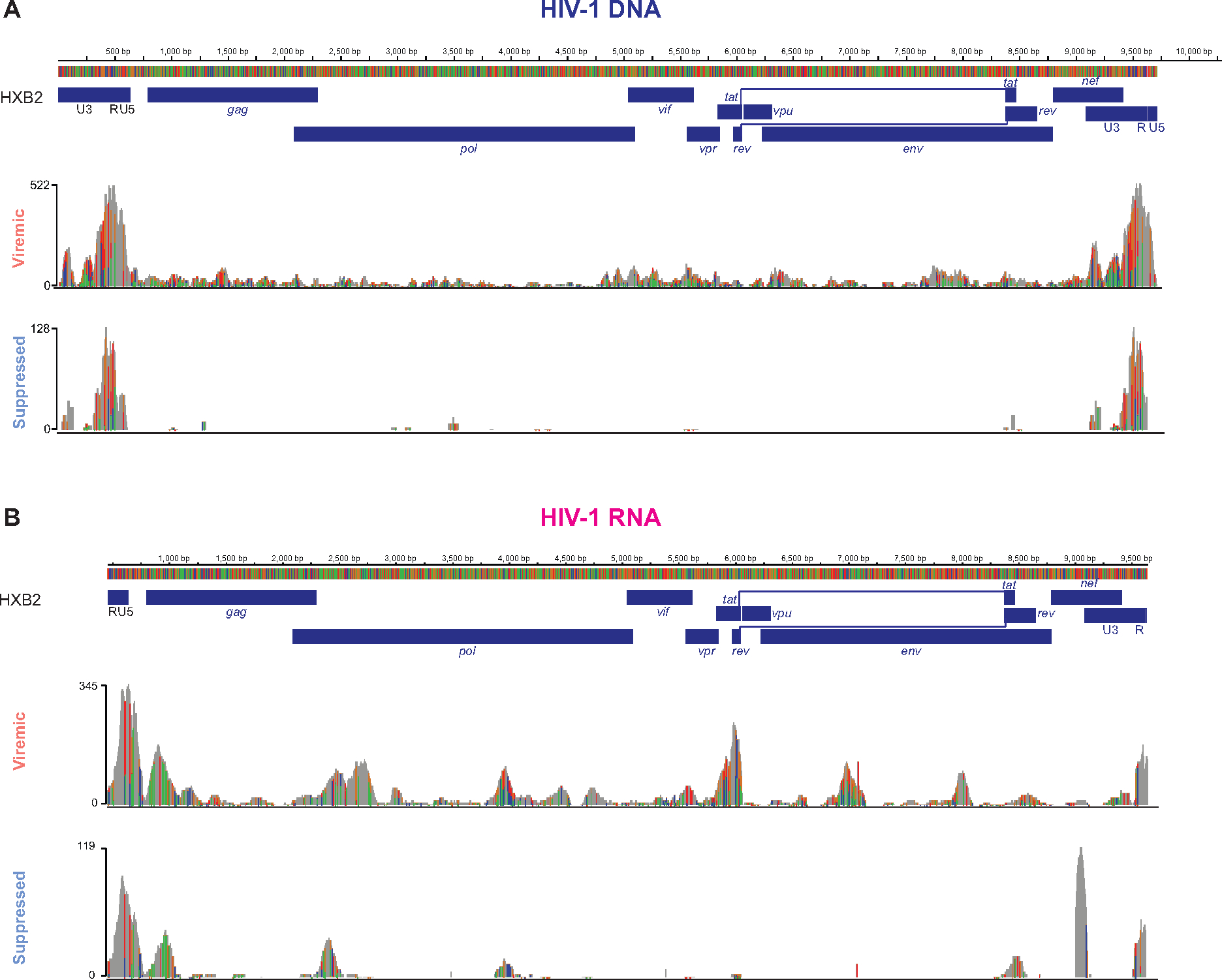
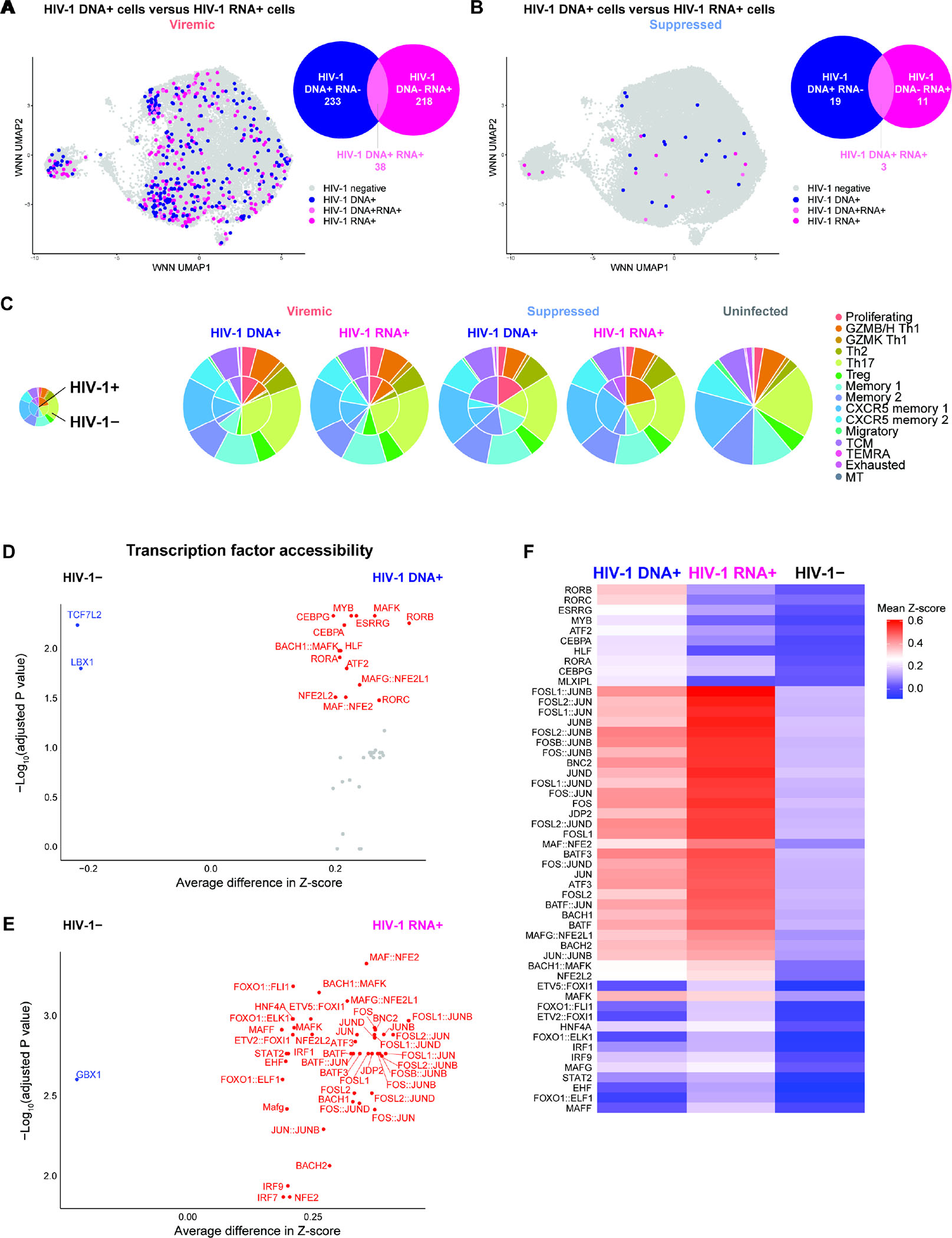
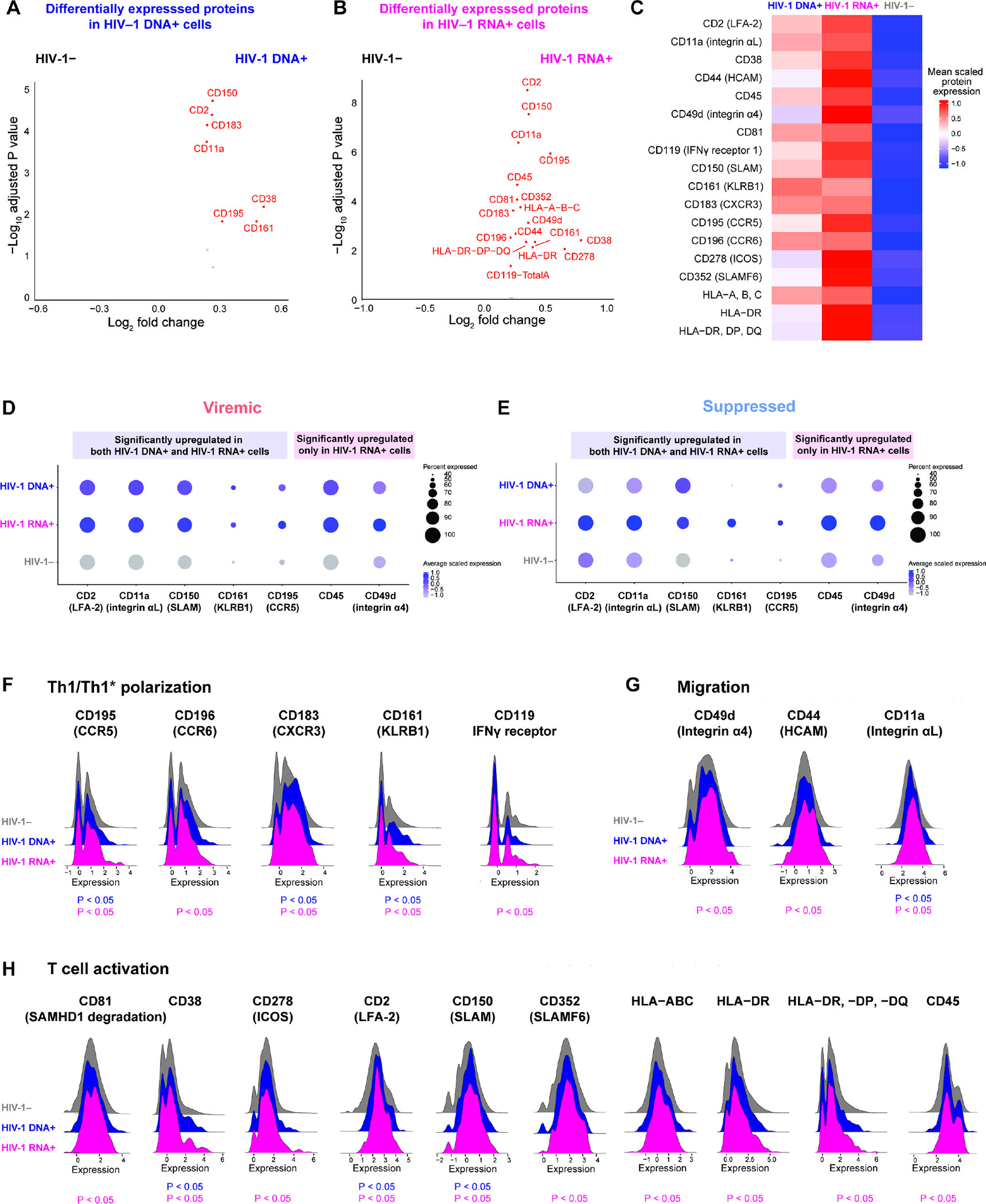
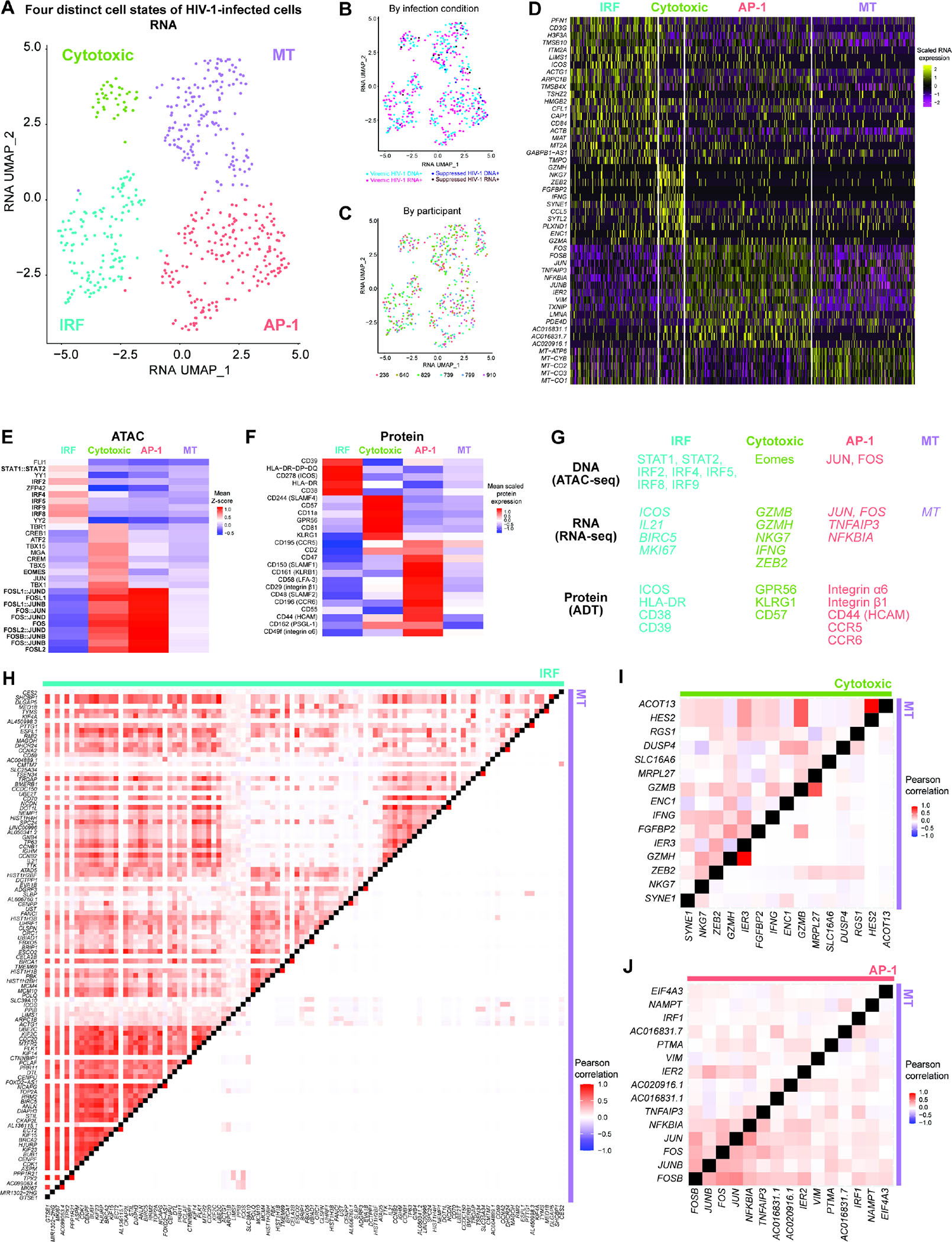
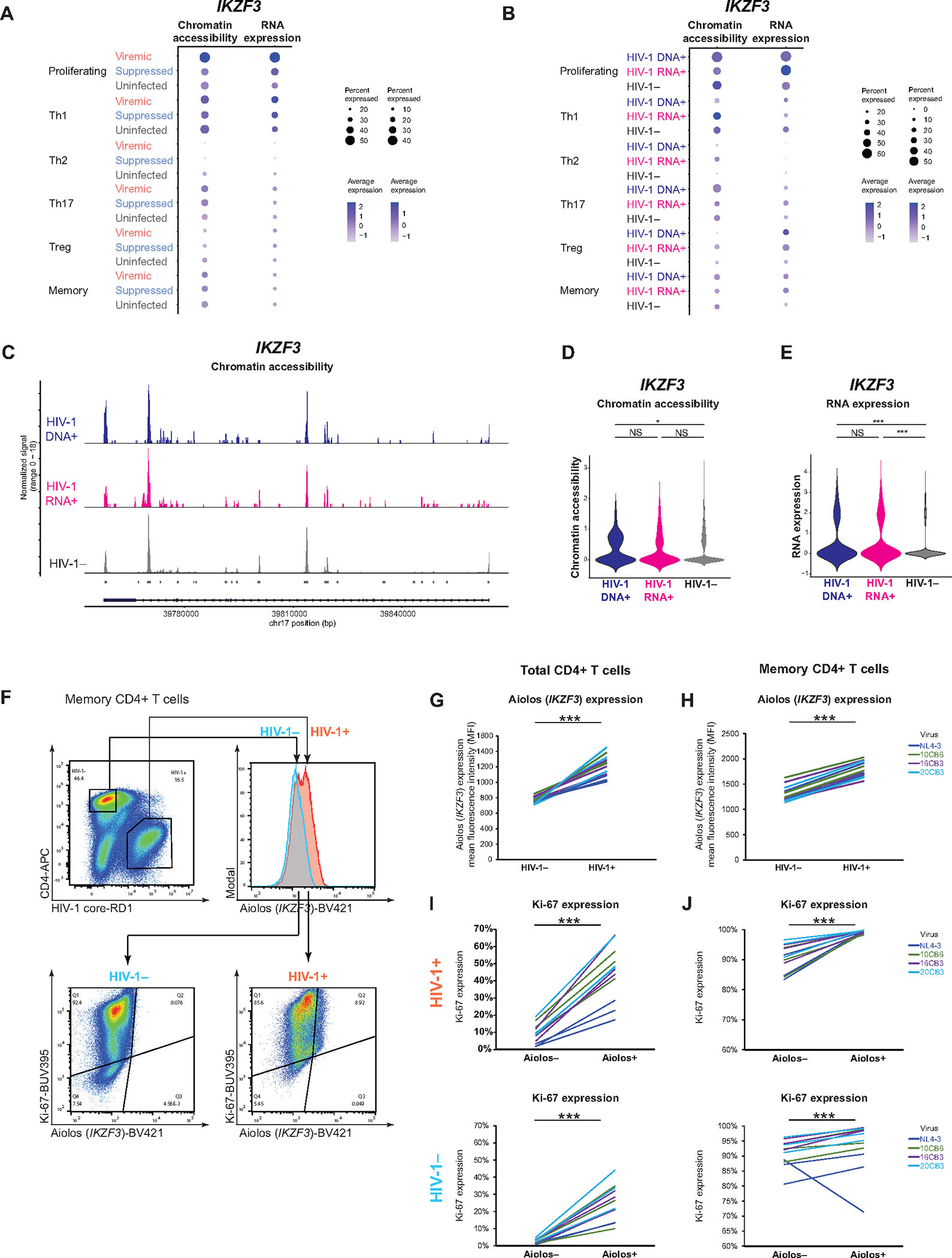
Understanding how HIV-1-infected cells proliferate and persist is key to HIV-1 eradication, but the heterogeneity and rarity of HIV-1-infected cells hamper mechanistic interrogations. Here, we used single-cell DOGMA-seq to simultaneously capture transcription factor accessibility, transcriptome, surface proteins, HIV-1 DNA, and HIV-1 RNA in memory CD4+ T cells from six people living with HIV-1 during viremia and after suppressive antiretroviral therapy. We identified increased transcription factor accessibility in latent HIV-1-infected cells (RORC) and transcriptionally active HIV-1-infected cells (interferon regulatory transcription factor [IRF] and activator protein 1 [AP-1]). A proliferation program (IKZF3, IL21, BIRC5, and MKI67 co-expression) promoted the survival of transcriptionally active HIV-1-infected cells. Both latent and transcriptionally active HIV-1-infected cells had increased IKZF3 (Aiolos) expression. Distinct epigenetic programs drove the heterogeneous cellular states of HIV-1-infected cells: IRF:activation, Eomes:cytotoxic effector differentiation, AP-1:migration, and cell death. Our study revealed the single-cell epigenetic, transcriptional, and protein states of latent and transcriptionally active HIV-1-infected cells and cellular programs promoting HIV-1 persistence.
Keywords: Aiolos; HIV cure; HIV latent reservoir; HIV persistence; IKZF3; T cell differentiation; acute viral infection; memory CD4(+) T cells; single-cell ATAC-seq; single-cell RNA-seq; transcription factor.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Wu VH, Nordin JML, Nguyen S, Joy J, Mampe F, Del Rio Estrada PM, Torres-Ruiz F, González-Navarro M, Luna-Villalobos YA, Ávila-Ríos S, et al. (2023). Profound phenotypic and epigenetic heterogeneity of the HIV-1-infected CD4(+) T cell reservoir. Nat Immunol 24, 359–370. 10.1038/s41590-022-01371-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DA051906/DA/NIDA NIH HHS/United States
- UM1 AI164565/AI/NIAID NIH HHS/United States
- UM1 DA051410/DA/NIDA NIH HHS/United States
- UM1 AI164570/AI/NIAID NIH HHS/United States
- R01 AI145164/AI/NIAID NIH HHS/United States
- R33 DA047037/DA/NIDA NIH HHS/United States
- U01 DA053628/DA/NIDA NIH HHS/United States
- U54 AI170856/AI/NIAID NIH HHS/United States
- P50 AI150464/AI/NIAID NIH HHS/United States
- R01 DA032106/DA/NIDA NIH HHS/United States
- P01 AI169768/AI/NIAID NIH HHS/United States
- R01 AI174863/AI/NIAID NIH HHS/United States
- R01 DA040532/DA/NIDA NIH HHS/United States
- UM1 AI126620/AI/NIAID NIH HHS/United States
- R61 DA047037/DA/NIDA NIH HHS/United States
- T32 AI055403/AI/NIAID NIH HHS/United States
- R01 AI141009/AI/NIAID NIH HHS/United States
- R37 AI147868/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
